
INTRODUCTION
Currently, drug design relies on a thorough understanding of biological targets and the structure–activity relationships of potential drug candidates. This approach integrates molecular biology, biochemistry, and computational chemistry to develop compounds that deliver the desired therapeutic effects while minimizing adverse side effects. Given that approximately 30% of drug candidates fail due to inadequate pharmacokinetic properties [1], it is essential to achieve a balanced pharmacodynamic and pharmacokinetic profile in drug-like molecules during the development of new pharmaceuticals. Consequently, a primary goal in drug discovery is to design molecules with high affinity for their targets along with suitable physicochemical properties. One effective strategy for discovering new drugs involves identifying promising structures.
A structure that has gained interest in recent years in drug design is norbornene and its derivatives. Homonorbornane/ene or heteronorbornane/ene (oxa or aza) skeletons are specialized systems found in natural compounds such as camphor, cantharidins, cineole, and their derivatives [2–4]. Due to their unique reactivity and fixed geometries, norbornenyl derivatives are highly valuable synthons—aptly described as “molecular LEGO”—for the preparation of molecules with defined stereochemistry and regiochemistry [2]. These systems are employed in specific reactions that involve bicyclic skeletons, such as nucleophilic or metathesis ring-opening reactions [5,6]. In the field of medicinal chemistry, norbornene and its derivatives have gained significant attention primarily due to their potential applications in cancer therapy [7]. These scaffolds have been shown to induce antitumor effects through various mechanisms, including the inhibition of estrogen receptors (ERs), androgen receptors (ARs), and carbonic anhydrase, as well as the regulation of proteins involved in apoptosis, autophagy, and the cell cycle [7].
Another key structural component of many anticancer drugs currently on the market is heterocycles, characterized by the replacement of a carbon atom in the ring structure with elements such as oxygen, nitrogen, or sulfur. Their anticancer activity stems from their ability to participate in diverse intermolecular interactions, including hydrogen bond donation and acceptance, π-stacking interactions, metal coordination, van der Waals forces, and hydrophobic interactions. These versatile binding capabilities enable heterocycles to interact with enzymes in various ways, effectively disrupting multiple biological pathways associated with cancer progression [8]. Piperazine is an organic compound characterized by a six-membered nitrogen heterocycle known as hexahydropyrazine, which contains two nitrogen atoms located at positions 1 and 4 [9]. This scaffold is recognized as a privileged structure in drug design [10]. Among the drugs approved by the US Food and Drug Administration, around 75% are nitrogen heterocycles, with piperazine ranking as the third most common structure [11]. These compounds serve a wide range of therapeutic purposes, including use as antipsychotics, antihistamines, antianginal agents, antidepressants, anticancer agents, antivirals, cardioprotective agents, anti-inflammatory drugs, and imaging agents [12]. The broad therapeutic applications of the piperazine scaffold arise from the observation that even minor modifications to the nucleus can result in significant variations in the medicinal potential of the resulting molecules. Furthermore, the presence of two nitrogen atoms with appropriate pKa values in the piperazine core enhances the pharmacokinetic profiles of drug candidates by improving the water solubility and thus their bioavailability [13,14]. As a result, the characteristics of the piperazine nucleus make it a valuable pharmacophore in the rational design of drugs.
Over the past two decades, the molecular hybridization of two or more pharmacophores into a single molecule gained popularity as a rational drug design strategy [15]. This hybrid approach in drug design is more than just combining two or more drugs into one formulation. Instead, it focuses on creating novel, more effective single drugs with better bioavailability and potentially reduced toxicity compared to the individual drug. The hybrid drug can be especially valuable in the treatment of multifactorial diseases such as cancer, neurological disorders, or infectious diseases [16–18].
Taking into account that hybridization of the privilege structures is one of the productive approaches in drug design, in the present study we carried out the synthesis of the piperazine derivatives containing the norbornenyl fragment through a straightforward pathway. All synthesized compounds were evaluated for their interaction with potential target proteins. Additionally, in silico assessment of the absorption, distribution, metabolism, excretion and toxicity (ADMET) profile and in vitro studies of cytotoxicity and genotoxicity of the compounds were performed.
MATERIALS AND METHODS
Synthesis
General information: Melting points were determined using a Buetus apparatus. 1H NMR and 13C NMR (300 MHz) spectra were recorded with dimethyl sulfoxide (DMSO)-d6 or DMSO-d6/CCl4 1/3 [Varian Mercury (300 MHz)]. The spectra are presented in the Supplementary Materials Figures S1 and S2. IR spectra were recorded using Nexus (Thermo Nicolet Corporation, USA). The results are presented in the Supplementary Materials Figure S3. Element analysis was performed using the Korshuna-Klimova apparatus. Mass spectra were recorded on XEVO G3 QTof spectrometer (Waters Corporation Company, Milford, Massachusetts), and results are presented in the Supplementary Materials Figures S4 and S5.
2,2’-((piperazine-1,4-dicarbonyl)bis(4,1-henylene))bis(3a,4,7,7a-tetrahydro-1H-4,7-methanoisoindole-1,3(2H)-dione) (4a).
The 5.1 g (0.031 mol) of anhydride (1) and 4.3 g (0.031 mol) of 4-aminobenzoic acid (2a) were heated at 175°C for 2 hours. The reaction mixture was washed with ethanol followed by diethyl ether and then filtered. The crystals of 3a were formed: 6.2 g (70%), mp of 221°C–222°C (lit. 224°C–228oC) [19]. The obtained acid (3a) (6.2 g, 0.022 mol) was heated with 2.6 g (0.022 mol) thionyl chloride in 10 ml benzene. The mixture was refluxed until the release of HCl and SO2 gasses was complete. The crystals were subjected to benzene addition and removal twice. Following this, the forming crystals were washed with benzene and then filtered. The yield of chloroanhydride is as follows: 5.7 g (86%), mp 145°C–146°C. The chloroanhydride (4.7 g, 0.0156 mol) of the corresponding acid was refluxed with piperazine (0.67 g, 0.0078 mol) in 20 ml benzene in the presence of triethylamine (2.3 g, 0.0227 mol) for 5 hours. The resulting salt was dissolved in water and then filtered. Yield: 4g, (83%), mp: 325°C–326°C. . IR, ν, cm−1: 1703, 176 (CONCO), 1653 (CON). NMR (300 MHz, DMSO-d6) 1H, δ: 1.61–1.68 m (4H, CH2), 3.32–3.37 m (4H, CH), 3.35–3.75 br (8H, NCH2), 3.49–3.53 m (4H, CH), 6.21–6.25 m (4H, CH=), 7.17–7.23 m (4H, C6H4), 7.46–7.52 m (4H, C6H4). 13C: 41.1 br (2*NCH2), 44.0 br (2*NCH2), 44.8 (4*CH), 45.4 (4*CH), 51.7 (2*CH2), 126.9 (4*CH=), 127.6 (4*CH=), 133.1 (2*C), 134.5 (4*CH), 135.3 (2*C), 168.4 (2* C=O), 176.5 (4* C=O). C36H32N4O6, Calculated %: C, 70.12; H, 5.23; N, 9.09. Found %: C, 70.28; H, 5.03; N, 8.95.
2,2’-(piperazine-1,4-diylbis(2-oxoethane-2,1-diyl))bis(3a,4,7,7a-tetrahydro-1H-4,7-methanoisoindole-1,3(2H)-dione) (4b).
To 5.3 g (0.0323 mol) anhydride (1), 2.4 g (0.032 mol) glycine (2b) was added, and the mixture was refluxed at 175°C for 45 minutes. The reaction mixture was washed with acetone to yield acid 3b: 5.1 g (72%), mp 144°C (lit.151°C–151.5°C) [20]. The obtained acid (3b) (4.9 g, 0.022 mol) was dissolved in 10 ml benzene, and 3 g (0.0252 mol) thionyl chloride was added. The mixture was refluxed until the release of HCl and SO2 gasses was complete. Benzene was removed under reduced pressure, and the residue was filtered to give the chloroanhydride: 4.5 g (85.6%), mp 261°C–262°C. The chloroanhydride (4.5 g, 0.019 mol) of the corresponding acid and 1 g (0.0116 mol) piperazine were refluxed in 20 ml benzene in the presence of 3.4 g (0.034 mol) triethylamine for 5 hours. Then, the residue was washed with a 1:1 mixture of water and ethanol, followed by ethanol alone, and then filtered. Yield: 2.85 g, (50%), mp: above 312°C–314°C. IR, ν, cm−1: 1708, 1764 (CONCO), 1673 (CON). NMR (300 MHz, DMSO-d6) 1H, δ: 1.53–1.62 m (4H, CH2), 3.23–3.29 m (4H, CH), 3.35–3.63 m (8H, C4H8N2), 3.40–3.45 m (4H, CH); 4.13 br (4H, NCH2), 6.01–6.07 m (4H, CH=). 13C: 38.7 (2*NCH2), 41.2 br (2*NCH2), 43.5 br (2*NCH2), 44.2 (4*CH), 45.4 (4*CH), 51.7 (2*CH2), 134.3 (4*CH=), 163.7 (2* C=O), 176.7 (4* C=O). C26H28N4O6, Calculated %: C, 63.40; H, 5.73; N, 11.38. Found %: C, 63.71; H, 5.37; N, 11.58. TOF MS ES+ [MH] m/z: Calculated for C26H28N4O6 493.2087, found 493.2090 (Supplementary Materials Fig. S3).
2,2’-(piperazine-1,4-diylbis(3-oxopropane-3,1-diyl))bis(3a,4,7,7a-tetrahydro-1H-4,7-methanoisoindole-1,3(2H)-dione) (4c).
To 4.2 g (0.0256 mol) anhydride (1), 2.4 g (0.0267 mol) β-alanine (2c) was added, and the mixture was refluxed in 20 ml toluene for 5 hours. The residue was filtered to yield acid 3c: 5.5 g (91%), mp 126°C–127°C (oily material) [21]. The obtained acid (3c) (5 g, 0.0213 mol) and 2.5 g (0.0210 mol) thionyl chloride were refluxed in 30 ml benzene until the release of HCl and SO2 gases was complete. The resulting chloroanhydride was obtained as 4 g (75%), mp 101°C–102°C. The chloroanhydride (4 g, 0.0157 mol) of the corresponding acid, 0.67 g (0.0078 mol) piperazine, and 3.1 g (0.031 mol) triethylamine were refluxed in 20 ml benzene for 5 hours. The residue was dissolved in water, filtered, recrystallized from an ethanol/dioxane mixture, and filtered again Yield: 1.6 g, (40%), mp: 253°C–255°C. IR, ν, cm−1: 1693, 1761 (CONCO), 1640 (CON). NMR (300 MHz, DMSO-d6) 1H, δ: 1.49–1.58 m (4H, CH2), 2.35–2.46 m (4H, CH2-CO), 3.20–3.26 m (4H, CH); 3.27–3.47 m (12H, NCH2); 3.30–3.34 m (4H, CH); 6.04–6.08 (4H, CH=). 13C: 30.3 (2*CH2), 33.8 (2*NCH2), 42.3 br (4*NCH2), 44.2 (4*CH), 45.1 (4*CH), 51.5 (2*CH2), 134.2 (4*CH=), 168.1 (2* C=O), 177.1 (4* C=O). C28H32N4O6, Calculated %: C, 64.60; H, 6.20; N, 10.76. Found %: C, 64.39; H, 6.53; N, 10.91.
2,2’-(piperazine-1,4-diylbis(1-oxopentane-1,2-diyl))bis(3a,4,7,7a-tetrahydro-1H-4,7-methanoisoindole-1,3(2H)-dione) (4d).
Anhydride (1) (6.5 g, 0.04 mol) and norvaline (2d) (4.7 g, 0.04 mol) were refluxed at 175°C for 45 minutes, Afterward, the reaction mixture was washed with acetone, yielding acid 3d (7.4 g, 70%), mp 135°C. The obtained acid (3d) (6.9 g, 0.0262 mol) was refluxed with 2.7 g (0.023 mol) thionyl chloride in 30 ml benzene until the release of HCl and SO2 gases was complete, providing 5.4 g (73%) of chloroanhydride, mp 170°C–171°C. We further refluxed the chloroanhydride (5.4 g, 0.019 mol) with 1.6 g (0.019 mol) piperazine in 20 ml benzene for 45 minutes and recrystallized the residue from an ethanol/dioxane mixture. Yield: 4.4 g, (40%), mp: 260°C–265°C. IR, ν, cm−1: 1702, 1776 (CONCO), 1654 (CON). NMR (300 MHz, DMSO-d6/CCl4 1/3) 1H, δ: 0.89 t (6H, J=7.3, CH3), 1.13–1.26 m (4H, CH2CH3), 1.59 d (2H, J=8.6, CHCH2CH); 1.67 d (2H, J=8.6, CHCH2CH); 1.70–1.91 m (4H, CH2CH2CH3), 3.13–3.48 br (8H, C4H8N2), 3.29–3.35 m (8H, CH); 4.52 dd (2H, J1=9.0, J2=5.7, NCH); 6.06–6.08 m (4H, =CH). 13C: 13.1 (2*CH3), 18.8 (2*CH2), 29.8 (2*CH2), 41.5 br (4*NCH2), 44.2 (4*CH), 44.71 (2*CH), 44.73 (2*CH), 50.4 (2*NCH), 51.6 (2*CH2), 134.1 (4*CH=), 165.8 (2* C=O), 175.8 (2* C=O), 175.9 (2* C=O). C32H40N4O6, Calculated %: C, 66.65; H, 6.99; N, 9.72. Found %: C, 66.81; H, 7.02; N, 9.51. TOF MS ES+ [MH] m/z: Calculated for C32H40N4O6 577.3026, found 577.3028 (Supplementary Materials Fig. S4).
2,2’-(piperazine-1,4-diylbis(1-oxohexane-2,1-diyl))bis(3a,4,7,7a-tetrahydro-1H-4,7-methanoisoindole-1,3(2H)-dione) (4e).
The 3.5 g (0.0213 mol) anhydride (1) and 2.8 g (0.0213 mol) norleucine (2e) were refluxed at 170°C for 45 minutes. The residue was filtered, yielding acid 3e: 4.1 g (69%), mp 185°C. The obtained acid (3e) (4.1 g, 0.0148 mol) was refluxed in 20 ml benzene with 1.8 g (0.0149 mol) thionyl chloride until the release of HCl and SO2 gases was complete. The residue was washed with benzene, yielding chloroanhydride: 3.5 g (80%), mp 200°C. We then refluxed the chloroanhydride (3.5 g, 0.0118 mol), 0.5 g (0.0058 mol) piperazine, and 2.3 g (0.0227 mol) triethylamine in 20 ml benzene for 5 hours. The resulting residue was dissolved in water, filtered, and recrystallized from ethanol. Yield: 1.8 g, (50%), mp: 240°C–241°C. IR, ν, cm−1: 1704, 1770 (CONCO), 1654 (CON). NMR (300 MHz, DMSO-d6) 1H, δ: 0.83 t (6H, J=7.1, CH3), 1.01–1.35 m (8H, CH2CH2CH3), 1.49–1.62 m (4H, CHCH2CH); 1.63–1.93 m (4H, NCHCH2), 3.07–3.55 br (8H, C4H8N2), 3.22–3.29 m (4H, CH); 3.37–3.41 m (4H, CH); 4.57 dd (2H, J1=9.0, J2=5.7, NCH); 6.04–6.09 m (4H, =CH). 13C: 13.7 (2*CH3), 21.6 (2*CH2), 27.6 br (2*CH2), 27.7 (2*CH2), 41.7 br (4*NCH2), 44.2 (4*CH), 44.86 (2*CH), 44.92 (2*CH), 50.7 br (2*NCH), 51.9 (2*CH2), 134.5 (4*CH=), 166.3 (2* C=O), 176.7 (2* C=O), 176.8 (2* C=O). C34H44N4O6, Calculated %: C, 67.94; H, 7.49; N, 9.05. Found %: C, 67.61; H, 7.77; N, 9.35.
2,2’-(piperazine-1,4-diylbis(1-oxo-3-phenylpropane-1,2-diyl))bis(3a,4,7,7a-tetrahydro-1H-4,7-methanoisoindole-1,3(2H)-dione) (4f).
The 4 g (0.0244 mol) anhydride (1) and 4.1 g (0.0246 mol) β-phenylalanine (2f) were refluxed in 20 ml p-xylene for 1 hour. The reaction mixture was filtered and dried, yielding acid 3f: 5.7 g (75%), mp 142°C (lit. 152°C–154oC) [21]. The obtained acid (3f) (5.7 g, 0.0183 mol) was refluxed in 30 ml benzene with 3 g (0.0252 mol) thionyl chloride until the release of HCl and SO2 gases was complete. This yielded the chloroanhydride: 5.4 g (89%), mp 190°C–192°C. The chloroanhydride (5.4 g, 0.0164 mol) was refluxed with 1.4 g (0.0164 mol) piperazine in 20 ml p-xylene for 6 hours. The resulting crystals were dissolved in water and recrystallized from an ethanol-benzene mixture. Yield: 9.2 g (83%), mp: 250°C–255°C. IR, ν, cm−1: 1702, 1776 (CONCO), 1654 (CON). NMR (300 MHz, DMSO-d6) 1H, δ: 1.41–1.47 m (4H, CH2), 3.08–3.20 m (8H, CH), 3.15–3.70 br (12H, NCHCH2, C4H8N2), 4.97–5.03 m (2H, NCH), 5.48–5.61 m (2H, =CH), 5.61–5.85 m (2H. =CH), 7.11–7.29 m (10H, C6H5). 13C: 33.6 (CH2), 33.7 (CH2), 41.5 br (4*NCH2), 44.1 (2*CH), 44.2 (2*CH), 44.8 (2*CH), 44.9 (2*CH), 51.5 (CH2), 51.6 (CH2), 51.65 (2*CH), 126.4 (2*CH=), 127.9 (4*CH), 129.2 (2*CH), 129.3 (2*CH=), 133.9 (2*CH=), 134.4 (2*CH=), 137.0, 137.02, 166.0 (2* C=O), 176.56 (2* C=O), 176.60 (2* C=O). C40H40N4O6, Calculated %: C, 71.41; H, 5.99; N, 8.33; Found %: C, 71.58; H, 6.10; N, 8.58.
2,2’-(((1E,1’E)-piperazine-1,4-diylbis(3-oxoprop-1-ene-3,1-diyl))bis(3,1-phenylene))bis(3a,4,7,7a-tetrahydro-1H-4,7-methanoisoindole-1,3(2H)-dione) (4g).
Anhydride (1) (4.1 g, 0.025 mol) and 3-(3-aminophenyl)acrylic acid (2g) (4.1 g, 0.025 mol) were refluxed at 175°C for 30 minutes. The reaction mixture was recrystallized from dioxane, yielding acid 3g: 6 g (77%), mp 280°C–283°C. The obtained acid (3g) (6 g, 0.0194 mol) was refluxed with 2.3 g (0.0194 mol) thionyl chloride in 20 ml toluene until the release of HCl and SO2 gases was complete. The reaction mixture was washed with diisopropyl ether, yielding chloroanhydride: 5.4 g (85%), mp 180°C. The chloroanhydride (3.5 g, 0.0106 mol) and 0.9 g (0.0106 mol) piperazine were refluxed in 20 ml boiling toluene for 4 hours. The resulting precipitate was dissolved in water, filtered, and recrystallized from a water/ethanol mixture. Yield, 3g (42%), mp: above 300°C–302°C. IR, ν, cm−1: 1705, 1770 (CONCO), 1658 (CON). NMR (300 MHz, DMSO-d6) 1H, δ: 1.57–1.65 m (4H, CH2), 3.32–3.41 m (4H, CH), 3.44–3.57 m (4H, CH), 3.57–3.84 br (8H, C4H8N2), 6.24–6.30 m (4H, =CH), 7.08–7.13 m (2H, C6H4), 7.21–7.31 m (2H, =CH); 7.41–7.52 m (2H, C6H4), 7.45–7.55 m (4H, Ar); 7.73–7.81 m (2H, C6H4). 13C: 41.5 br (4*NCH2), 44.7 (2*CH), 45.4 (2*CH), 51.6 (2*CH2), 119.2 (2*CH=), 126.6 (2*CH=), 127.7 (2*CH), 128.1 (2*CH), 129.2 (2*CH), 132.7 (2*C), 134.5 (4*CH=), 135.9 (2*C), 140.5 (2*CH), 164.4 (2* C=O), 176.5 (4* C=O). C40H36N4O6, Calculated %, C, 71.84; H, 5.43; N, 8.38; Found %, C, 71.95; H, 5.21; N, 8.51.
2,2’-(piperazine-1,4-diylbis(3-methyl-1-oxobutane-1,2-diyl))bis(3a,4,7,7a-tetrahydro-1H-4,7-methanoisoindole-1 ,3-(2H)-dione) (4h).
Anhydride (1) (4 g, 0.0244 mol) and valine (2h) (2.86 g, 0.0244 mol) were refluxed in 25 ml p-xylene. The reaction mixture was washed with diethyl ether, yielding acid 3h: 3.6 g (56%), mp 94°C–95°C (lit. 107°C–109oC) [21]. The obtained acid (3h) (3.6 g, 0.0137 mol) was refluxed with 1.63 g (0.0137 mol) thionyl chloride until the release of HCl and SO2 gases was complete. This yielded the chloroanhydride: 3.6 g (93%), mp 94°C–95°C. The chloroanhydride (3.6 g, 0.0128 mol) and 1.1 g (0.0128 mol) piperazine were refluxed in 20 ml benzene for 4 hours. The resulting precipitate was dissolved in a small amount of water, recrystallized from a dioxane-water mixture, and washed with diethyl ether. Yield: 4.7 g, (64%) mp: 225°C–226°C. IR, ν, cm−1: 1707, 1768 (CONCO), 1648 (CON). NMR (300 MHz, DMSO-d6) 1H, δ: 0.72 d (6H, J=7.0, CH3), 0.84 d (6H, J=6.5, CH3), 1.48–1.61 m (4H, CH2), 2.52–2.71 m (2H, CH(CH3)2), 3.01–3.67 m (8H, C4H8N2); 3.22–3.28 (4H, CH); 3.37–3.43 (4H, CH); 4.26–4.42 m (2H, NCH), 5.98–6.22 m (4H, =CH). 13C: 18.8 (2*CH3), 20.0 (2*CH3), 26.55 (CH), 26.57 (CH), 41.4 br (2*NCH2), 41.7 br (2*NCH2). 44.3 (4*CH), 44.91 (2*CH), 44.99 (2*CH), 52.0 (2*CH2), 55.5 (2*NCH), 134.6 (2*CH=), 134.8 (2*CH=), 165.6 br (2* C=O), 176.8 (2* C=O), 176.9 (2* C=O). C32H40N4O6, Calculated %: C, 66.65; H, 6.99; N, 9.72. Found %: C, 66.78; H, 6.81; N, 9.61.
Computational Studies
For the assessment of potential interactions of norbornenylpiperazine compounds with the target proteins, the Docking to ligand Field Activity prediction Model (DFA) implemented in ICM-Pro 3.8-3. molecular modeling software (MolSoft LLC, San Diego, CA) was used. This involved applying the hybrid 4D/2D model from MolScreen [22] combines flexible docking with ligand-based activity prediction to estimate ligand binding affinities and biological activities. The approach integrates molecular interaction fields from a quantitative structure-activity relationship-like model with docking scores to account for both structural and physicochemical properties of ligands. Default software parameters were applied, and the molpKd score was used to evaluate ligand–protein interactions. A molpKd score greater than six signifies an interaction at submicromolar concentrations. The Toxometris.ai platform (https://portal.toxometris.ai) was used to calculate physicochemical descriptors and predict the ADMET properties and drug-likeness of the compounds. The SMILES representations of compounds were generated and subsequently converted to canonical SMILES using RDKit software [23].
Cell Culture
The Michigan Cancer Foundation (MCF)-7 human breast cancer cells were maintained at 37°C in a cell incubator, with a humid atmosphere and 5% CO2. The cells were cultured in T-flasks, containing Dulbecco’s modified Eagle’s medium F12 (DMEM F12), supplemented with Phenol Red, 10% fetal bovine serum (FBS; Sigma Aldrich, USA), 2 mM L-glutamine and 100 mg/ml penicillin and 100 mg/ml streptomycin (Sigma Aldrich, USA).
Cell Viability Assay
The cytotoxicity of norbornenylpiperazine compounds was evaluated by the MTT method. For this, the MCF-7 human breast cancer cells were seeded at the density of 0.15 × 105 per well in a 96-well plate and cultivated for 48 hours in DMEM, supplemented with 10% FBS (Sigma Aldrich, USA), 2 mM L-glutamine and 100 mg/ml penicillin and 100 mg/ml streptomycin (Sigma Aldrich, USA) at 37°C and 5% CO2. The stock solutions of compounds were prepared in 100% DMSO based on the highest soluble concentration with a final concentration of 2.5% DMSO in each well (Table 1). The cell’s viability was evaluated following a 48-hour treatment. DMSO at the concentration of 2.5% was used as a control. MTT solution was added to each well, and after 3-hour incubation, DMSO was added to solubilize formazan. The absorbance was measured at 450 nm by a microplate reader (HumaReader HS 16670).
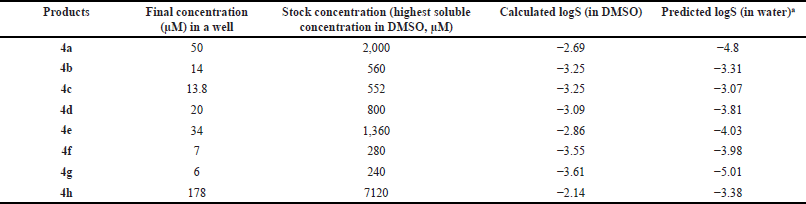 | Table 1. Concentrations of norbornenylpiperazine used in cytotoxicity assay. [Click here to view] |
Comet Assay
The comet assay was performed using the method described by Kumar et al. [24] (2015) with slight modifications. Briefly, after 48 hours of incubation with the compound’s highest soluble concentrations (Table 1) and two times lower, DMSO (2.5%) and 10 μM methyl methanesulfonate (MMS) control and cell pellets obtained through centrifugation were resuspended in PBS. A 70 μl volume of 0.5% low melting point agarose was mixed with 40 μl of the cell suspension, and the mixture was pipetted 45 μl onto half of the slides coated with 1% normal melting point agarose. Next, cover slips (22 × 22 mm) were placed on top of the slides and stored at 4°C. The slides were then immersed in a lysis solution (2.23 M NaCl, 0.09 M EDTA 0.009 M Tris, pH 10, 10% DMSO, 1% Triton X-100) for at least 1 hour at 4°C in the dark and subsequently soaked in electrophoresis buffer (1 mM EDTA pH 10, 0.3 M NaOH) for 20 minutes. Electrophoresis was conducted for 20 minutes at 25 V and 300 mA at 4°C. Following electrophoresis, the slides were immediately neutralized with 0.4 M Tris buffer (pH 7.5) for 5 minutes. The slides were stained with 20 μg/ml ethidium bromide (60 μl per slide) for at least 1 hour at 4°C in the dark. Images were captured using a fluorescence microscope (iScope 3153-PLi/6, Euromex, China). A total of 100 cells (50 cells from each of two halves of the slides) were randomly selected and analyzed using CaspLab software (https://casplab-comet-assay-software-project.soft112.com/). The DNA damage was quantified using the olive tail moment (OTM), a parameter calculated by multiplying the comet tail’s intensity by the distance it migrates from the center of the comet head.
Statistical Analysis
All experiments were performed in triplicates, and the data are presented as the mean ± SE of three independent experiments. Statistical analysis was conducted using GraphPad Prism version 8.0 (Boston, Massachusetts). To evaluate differences between treatments, the non-parametric Mann–Whitney U test was applied. The level of < 0.05 was considered statistically significant.
RESULTS AND DISCUSSION
Synthesis
The general strategy for the synthesis of the target compounds (Table 1) is summarized in Scheme 1. Briefly, 5-norbornene-2,3-dicarboxylic anhydride (1) reacted with amino acids (2a-h), under reflux to give the corresponding R-substituted carboxylic acids (3a-h), which reacted with thionyl chloride (SOCl2) to afford acyl chlorides of corresponding acids (4a-h). Subsequent interaction with piperazine with the appropriate R-substituted acyl chlorides performed in benzene or toluene with triethylamine, under reflux, provided the corresponding norbornenylpiperazine compounds (4a-h). The purification of the reaction mixture was carried out by crystallization from the appropriate solvent. All new compounds were characterized by 1H NMR, 13C NMR, and IR spectroscopy. It sounds like an efficient and streamlined approach—reaching final compounds without prolonged heating and with easy purification is a significant advantage in synthesis. Indeed, leveraging such strategies can streamline the process of identifying and synthesizing key scaffolds, which are crucial in rational drug design.
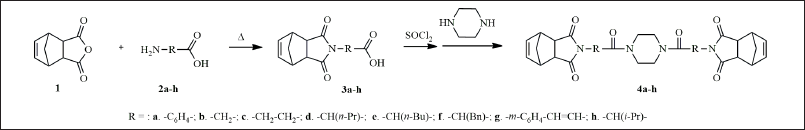 | Scheme 1. The general strategy for the synthesis of norbornenylpiperazine compounds (4a-h). [Click here to view] |
Prediction of Activity, Drug likeness, and ADMET properties
To assess the potential interactions of norbornenylpiperazine compounds with cancer therapeutic targets, the DFA model in MolScreen was applied. Analysis of molpKd values (Table 2) revealed that compound 4a likely interacts with the AR, compound 4b with peroxisome proliferator-activated receptor gamma (PPARγ), and compound 4f with the progesterone receptor (PR) at submicromolar levels. The other synthesized compounds exhibited potential interactions with the glucocorticoid receptor (GR).
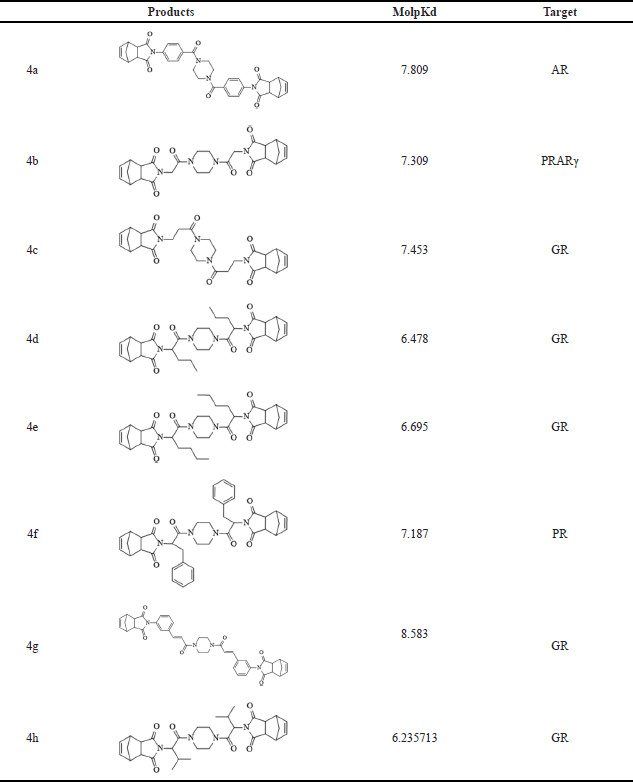 | Table 2. Prediction of potential interaction of studied compounds with receptors by the DFA model. [Click here to view] |
Accumulating evidence suggests the involvement of steroid nuclear receptors such as AR, PR, and GR in the progression and development of various cancers, making them appealing therapeutic targets. Drugs that inhibit AR are widely used as first-line therapies for metastatic prostate cancer. Additionally, the role of AR activation has garnered increasing recognition in the development and progression of breast cancer, positioning it as an attractive pharmaceutical candidate [25,26]. Recent research has underscored the role of PR signaling in the carcinogenesis of various gynecological cancers, including endometrial and ovarian cancers, as well as breast cancer [27–29]. Moreover, the efficacy of PR inhibition has been demonstrated in several clinical trials [30,31]; however, the effectiveness of these treatments is often limited by safety concerns, particularly the hepatotoxicity associated with some PR antagonists [32,33]. Studies on the biological effects of GR revealed its multifaceted role in breast cancer progression as it varies by subtype. High GR expression is associated with a favorable prognosis in ERα-positive breast cancers, whereas it promotes tumor metastasis in triple-negative breast cancers, highlighting GR as a promising therapeutic target for this subtype [34–37].
Overall, activity prediction revealed that some norbornenylpiperazine compounds may serve as promising candidates for targeting specific receptors involved in cancer therapy.
None of the synthesized norbornenylpiperazine violate Lipinski’ s rule of 5 (MW ≤ 500; logP ≤ 5; Hacc ≤ 10; Hdon ≤ 5) and/or Pfizer rule (logP > 3; TPSA < 75), which identifies the compounds as viable drug candidates (Supplementary Materials Table S1.). All of the compounds have a PAINS value of 0. PAINS (Pan-assay interference compounds) comprise classes of compounds defined by a common substructural motif that encodes for an increased chance of any member registering as a hit in any given assay that may be independent of platform technology. Such compounds are concomitantly less likely to be optimizable toward a useful compound. A common reason why PAINS register as hits in assays is that the substructure can confer an ability to interfere in biochemical assays [38]. So, the PAINS value of 0 demonstrates a low tendency of synthesized norbornenylpiperazine to nonspecifically interact with the proteome.
Evaluation of ADMET Properties Predictions
The ADMET properties of the norbornenylpiperazine compounds were predicted using the Toxometris-ADMET-Suite application. The complete list of predictions is available in Supplementary Materials Table S1.
The drug-like properties of synthesized norbornenylpiperazine compounds were analyzed based on selected ADME properties predictions. The prediction score was calculated based on the predicted value’s alignment within the acceptable range established for potential pharmaceuticals (Fig. 1). The Human Intestinal Absorption and Caco-2 permeability predictions provide valuable insights into the ability of chemicals to penetrate intestinal barriers, which is crucial for assessing oral bioavailability [39]. From this perspective, all evaluated compounds displayed promising results, with compounds 4b, 4f, and 4h exhibiting very good bioavailability, while others demonstrated moderate absorption potential. Yet, the predicted logS values ranged from −3.07 to −5.01, suggesting low aqueous solubility for the compounds, which was later confirmed experimentally (Table 1). However, the scoring function placed these compounds in the moderate to good range, as their logS values fall within the typical range for most commercially available drugs, which have logS values between −5 and 1 [40]. All compounds demonstrated a moderate to low probability of penetrating the blood–brain barrier. The compounds were predicted to have good plasma protein binding properties, indicating a potential for an extended half-life and sustained therapeutic effects (Fig. 1). However, three compounds (4e, 4f, and 4g) were predicted to exhibit poor microsomal stability characteristics, which suggests a higher likelihood of rapid metabolic degradation.
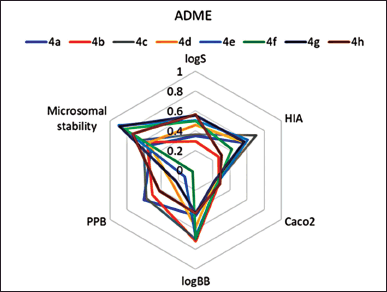 | Figure 1. Prediction scores for ADME properties, estimated based on in silico prediction values for synthesized norbornenylpiperazine compounds. A lower score indicates a higher likelihood that the compounds fall within the desirable ranges established for pharmaceuticals. [Click here to view] |
The toxicity profile of the synthesized compounds was evaluated in silico with respect to acute oral toxicity (AOT), hepatotoxicity, nephrotoxicity, and cardiotoxicity. According to AOT model predictions, all compounds, except for compound 4h, were classified as non-toxic (LD50 > 2000 mg/kg). The cardiotoxicity of the tested compounds was assessed through predicted hERG (human ether-à-go-go-related gene) potassium channel inhibition, which can disrupt the cardiac repolarization process, leading to prolonged action potentials and QT interval [41]. The predictions indicate a low probability of hERG inhibition for all evaluated compounds, suggesting that they are unlikely to exhibit cardiotoxic effects (Fig. 2a). The neurotoxicity of the compounds was assessed based on the predicted inhibition of acetylcholinesterase (AChE), an enzyme crucial for neurotransmitter regulation in the nervous system and a known marker of neurotoxicity [42]. Compounds 4c, 4a, 4d, 4f, and 4h exhibited a moderate probability of neurotoxicity (IC50 < 10 μM), while compounds 4b, 4e, and 4g demonstrated a non-neurotoxic profile probability (IC50 ≥ 10 μM). All compounds exhibited moderate probabilities of hepatotoxicity and moderate to high probabilities of nephrotoxicity (Fig. 2a). Although piperazine scaffolds and norbornene derivatives are favorable structures in drug design, including anticancer research, the increased risk of hepatotoxicity, neurotoxicity, and nephrotoxicity of studied compounds may be attributed to the presence of both 5-norbornene-2,3-dicarboximide and piperazine groups. Known piperazine derivatives drugs are primarily metabolized by cytochrome P450 with subsequent possible glucuronidation and/or sulfation [43]. Some of these derivatives produce reactive metabolites that can induce liver damage. For example, piperazine-containing drugs such as fluconazole have been reported to cause hepatotoxicity [44]. Some imide-containing compounds undergo metabolic activation in the liver, potentially leading to the formation of reactive intermediates [45]. Certain piperazine derivatives (indinavir, ciprofloxacin) are known to precipitate out in the renal tubules, leading to crystal-induced nephritis or acute kidney injury [46]. The norbornene structure is a strained bicyclic system that, when combined with piperazine, might lead to CNS excitation or sedation through poorly understood mechanisms. Norbornene derivatives have acute neurotoxic signs, including tremors, ataxia, and convulsions [47]. The specific toxicity would depend on how these compounds are metabolized. Reducing the toxic effects of norbornenylpiperazine derivatives can be approached through structural modifications. For example, it is possible to modify the chemical structure and introduce bulky groups around highly reactive functional groups to avoid metabolic pathways that produce toxic reactive intermediates.
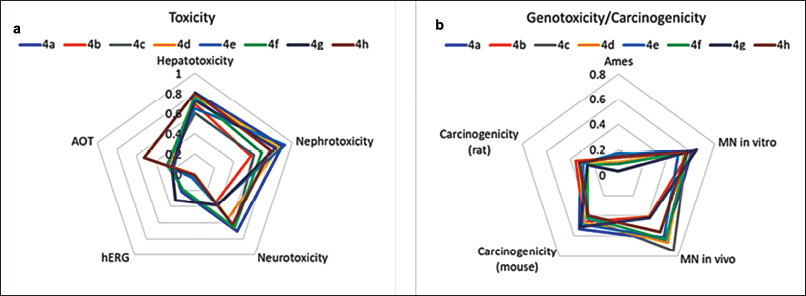 | Figure 2. Prediction scores for AOT and organ-specific toxicity endpoints (a) and genotoxicity/carcinogenicity endpoints (b), estimated based on in silico prediction values (Toxometris-ADMET-Suite) for synthesized norbornenylpiperazine compounds. A lower score indicates a reduced risk of toxicity. [Click here to view] |
While anticancer drugs are designed to treat existing cancers, some can increase the risk of secondary cancers due to their DNA-reactive nature. Many traditional chemotherapeutic agents, such as alkylating agents and topoisomerase inhibitors, cause DNA damage in both cancerous and healthy cells, which can lead to mutations and secondary malignancies [48]. However, modern approaches, such as targeting specific receptors (e.g., AR, GR, or PPARγ receptors), offer more precise mechanisms of action. These targeted therapies are specifically designed to minimize off-target effects and the carcinogenicity risk of new entities developed to target specific receptors must still be thoroughly evaluated to ensure long-term safety (ICH S1A, S1B) [49–51]. A set of mutagenicity (Ames test), genotoxicity (micronucleus in vitro and in vivo), and carcinogenicity (rats and mice) endpoints has been predicted to assess the potential carcinogenic risk of synthesized norbornenylpiperazines. Based on the predictions and the reliability scores for each, an overall score has been calculated to summarize the results and compare the potential risk of all compounds (Fig. 2b). The mutagenicity profiles of all compounds, as assessed by the Ames test, indicate a low risk of positive results; other endpoints revealed a moderate risk of positivity. The cumulative score, representing the average of all scores obtained from various endpoints, falls within the range of 0.37 to 0.45, indicating a transition from low to moderate risk for genotoxicity and/or carcinogenicity. It is known that the experimental values for these endpoints, upon which the predictive models are based, are often overestimated [52]. Given that toxicity assessment tests typically utilize very high doses to evaluate potential risk, it can be concluded that a careful dose selection approach would likely reveal a low risk of carcinogenesis for these compounds.
So, the synthesized norbornenylpiperazine compounds show promising drug-like properties, particularly in terms of bioavailability and low cardiotoxicity, making them potential candidates for further development. While solubility and some toxicological concerns, such as moderate to high risk of hepatotoxicity and nephrotoxicity, were predicted, these challenges can be addressed through careful dose selection and structural modifications. Overall, the moderate to low genotoxicity and carcinogenicity risk, combined with favorable ADME profiles, suggest that these compounds have potential for therapeutic use, provided that further optimization and experimental validation are carried out.
Cell Viability Assay
Eight norbornenylpiperazine compounds were screened for their cytotoxic activity against the MCF 7 breast cancer cell line using the MTT assay. This cell line was chosen as a model since it is characterized by the expression of classical nuclear receptors, including ER, AR, PR, and GR [53]. The IC50 values of the compounds could not be determined, as cell viability remained above 50% even at the highest soluble concentration tested. Therefore, only the effects at this highest soluble concentration were determined. According to the results (Fig. 3), compounds 4a, 4e, and 4h exhibited significant cytotoxicity against MCF-7 cells. However, it is important to note that the concentration of compound 4a used for treatment was quite high (50 μM) compared to the other compounds. The compound 4a was predicted to bind the AR with high affinity. This receptor plays a critical role in regulating gene expression related to cell growth and proliferation, particularly in androgen-dependent cancers. Targeting the AR, either as a monotherapy or in combination with conventional treatments, has proven to be effective for prostate cancer treatment and is increasingly being explored as a therapeutic approach in breast cancer [25]. It is less potent in inhibiting cell proliferation in AR-positive cell lines compared to Enzalutamide, a well-known AR ligand used in cancer treatment, which demonstrated 50% inhibition of the MCF-7 cell line at a concentration of 20 μM [54]. Although Enzalutamide is an effective treatment for prostate cancer, it is associated with several unfavorable parameters, including cardiotoxicity, bioavailability, and selectivity [55]. Therefore, further studies on compound 4a are necessary to accurately assess its comparative efficacy and safety. Nonetheless, compound 4a should still be considered a potential candidate for development as an AR ligand pharmaceutical.
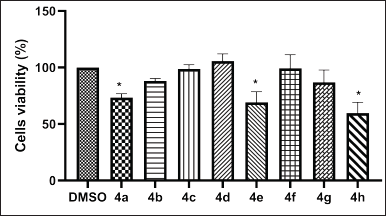 | Figure 3. In vitro cytotoxic activities of norbornenylpiperazine compounds 4a–h on the MCF-7 cell lines following a 48-h treatment. Data are presented as mean ± SE of three independent experiments. *p < 0.05 (Mann–Whitney U test). [Click here to view] |
Furthermore, compounds 4e and 4h, which have been found to interact with the GR, induced a significant inhibition of MCF-7 cell viability. GR-mediated drugs play a pivotal role in cancer treatment, primarily due to their anti-inflammatory and immunosuppressive properties. Additionally, these agents exert direct effects on cancer cells that express GR, influencing tumor growth and survival pathways [56]. The two most well-known GR-mediated drugs are dexamethasone and prednisone, which are widely used in the treatment of certain cancers [57]. The direct effects of dexamethasone on the MCF-7 cell line demonstrated up to 30% viability inhibition at a concentration of 0.1 μM [58], whereas the IC50 of prednisone was found to be greater than 30 μM [59] after 72 hours of treatment. In this study, we observed results comparable to prednisone, with cell viability decreasing by up to 69% for the 34 μM concentration of compound 4e and by 53% for the 180 μM concentration of compound 4h after 48 hours of treatment. Our findings are also consistent with those reported by Tieszen et al. [60], which indicates that GR inhibition leads to MCF-7 cell death independently of PR inhibition.
In conclusion, the screening of eight norbornenylpiperazine compounds revealed that compounds 4a, 4e, and 4h present promising avenues for further research, indicating potential as selective AR and/or GR ligands. Further studies are needed to refine the potency and selectivity of these compounds.
Genotoxic Activity
The genotoxic potential of the synthesized compounds was assessed using single cell gel electrophoresis (alkaline comet assay), which detects both single- and double-strand DNA breaks. This assay is highly sensitive to chemically induced DNA damage, capturing both repairable and non-repairable lesions. We selected this method to identify any potential DNA reactivity of the tested compounds. Compared to the micronucleus assay, which identifies strictly genotoxic compounds capable of transmitting damage through cell generations, the comet assay provides a broader detection of DNA damage [61]. DNA damage was assessed at two concentrations for each compound: a high concentration, corresponding to the maximum soluble dose used in cytotoxicity studies, and a low concentration, where no cytotoxicity was observed for any of the compounds. This was done to minimize false positive results related to cytotoxicity, as it is recommended that cell viability in the in vitro comet assay should be above 70%–75% [62].
As shown in Figures 4 and 5, treatment of MCF-7 cells with MMS (positive control) significantly increased the level of DNA damages compared to the vehicle control, with OTM values of 48.6 and 11.06, respectively. Among the norbornenylpiperazine compounds studied, none exhibited genotoxic potential at low concentrations, with DNA damage levels being equal to or even lower than those of the vehicle control. In contrast, compounds 4e, 4g, and 4h caused a significant increase in DNA damage at high concentrations, with OTM values of 143.5, 68.6, and 101.3, respectively, compared to the vehicle control and the positive control. Since these DNA damages were observed at high cytotoxic concentrations, where 30% to 50% inhibition of cell viability occurred, they can be attributed to the cytotoxic effects of the compounds and serve as evidence of cytotoxicity in the corresponding experiments, rather than genotoxicity. So, compounds that showed no cytotoxicity at either high or low concentrations were also non-genotoxic. In contrast, compounds that were cytotoxic at high concentrations exhibited increased levels of DNA damage. The only exception was compound 4a, which did not show any signs of genotoxicity at either low, non-cytotoxic concentrations or high, cytotoxic concentrations. Since compound 4a has been predicted to be a potential AR ligand, its behavior may be explained by AR-mediated cell death, particularly in the context of cancer. This process can involve pathways such as mitochondrial dysfunction and metabolic disturbances, which may lead to the inhibition of cell proliferation or promote apoptosis without observable signs of extensive DNA damages [63].
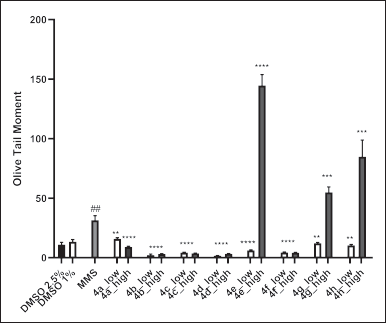 | Figure 4. In vitro genotoxic activities of norbornenylpiperazine compounds on the MCF-7 cell lines. Data are presented as means of olive tail moment ± SE of three independent experiments. **p < 0.01, ****p < 0.000 compared to MMS, ##p < 0.01 compared to negative DMSO controls (Mann–Whitney U test). [Click here to view] |
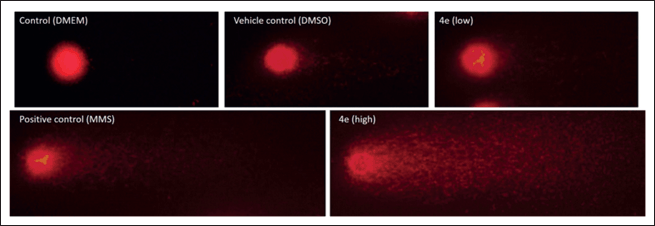 | Figure 5. Representative comet assay images: control (DMEM culture media), vehicle control (2.5% DMSO), positive control (MMS), and treatment with compound 4e at non-cytotoxic (17 μM) and cytotoxic (34 μM) concentrations for 48 hours. [Click here to view] |
It should be noted that these compounds were initially predicted to have low to moderate genotoxic profiles, based on assays other than the comet assay. Any discrepancies in the genotoxicity probabilities can be attributed to the fact that different assays evaluate various aspects of genetic toxicity; while they are complementary, they may yield divergent results. Considering both the in silico and in vitro studies, it can be concluded that the synthesized norbornenylpiperazine compounds have a low probability of being genotoxic.
CONCLUSIONS
In conclusion, the synthesized norbornenylpiperazine compounds displayed strong potential for interacting with cancer-related targets, including AR and GR. The in silico ADMET assessments revealed favorable pharmacokinetic properties with moderate toxicity profiles, while in vitro evaluations showed significant inhibition of MCF-7 breast cancer cell proliferation by compounds 4a, 4e, and 4h. Combining the in silico predictions with in vitro findings, the synthesized norbornenylpiperazine compounds demonstrate a low likelihood of genotoxicity. These findings establish a basis for the further development and functional evaluation of the selected synthesized norbornenylpiperazine compounds.
AUTHOR CONTRIBUTION
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
FINANCIAL SUPPORT
This work was supported by the Science Committee of RA (Research project ? 23LCG-1F002).
CONFLICTS OF INTERERST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All the data is available with the authors and shall be provided upon request.
PUBLISHER’S NOTE
All claims expressed in this article are solely those of the authors and do not necessarily represent those of the publisher, the editors and the reviewers. This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
USE OF ARTIFICIAL INTELLIGENCE (AI)-ASSISTED TECHNOLOGY
The authors declares that they have not used artificial intelligence (AI)-tools for writing and editing of the manuscript, and no images were manipulated using AI.
REFERENCES
1. Avdeef A. Physicochemical profiling (solubility, permeability and charge state). Curr Top Med Chem. 2001;1(4):277–351. doi: CrossRef
2. Vogel P, Cossy J, Plumet J, Arjona O. Derivatives of 7-oxabicyclo[2.2.1]heptane in nature and as useful synthetic intermediates. Tetrahedron. 1999;55(48):13521–642. doi: CrossRef
3. Spande TF, Garraffo HM, Edwards MW, Yeh HJC, Pannell L, Daly JW. Epibatidine: a novel (chloropyridyl)azabicyclo heptane with potent analgesic activity from an Ecuadoran poison frog. J Am Chem Soc. 1992;114(9):3475–78. doi: CrossRef
4. Chang LL, Truong Q, Doss GA, MacCoss M, Lyons K, McCauley E, et al. Highly constrained bicyclic VLA-4 antagonists. Bioorg Med Chem Lett. 2007;7(3):597–601. doi: CrossRef
5. Lautens M, Han W. Divergent selectivity in MgI2-mediated ring expansions of methylenecyclopropyl amides and imides. J Am Chem Soc. 2002;124(22):6312–16. doi: CrossRef
6. Arjona O, Csákÿ AG, Plumet J. Sequential metathesis in oxa- and azanorbornene derivatives. Eur J Org Chem. 2003;2003(4):611–22. doi: CrossRef
7. Calvo-Martín G, Plano D, Martínez-Sáez N, Aydillo C, Moreno E, Espuelas S, et al. Norbornene and related structures as scaffolds in the search for new cancer treatments. Pharmaceuticals (Basel). 2022;15(12):1465. doi: CrossRef
8. Hossain M, Habib I, Singha K, Kumar A. FDA-approved heterocyclic molecules for cancer treatment: synthesis, dosage, mechanism of action and their adverse effect. Heliyon. 2023;10(1):e23172. doi: CrossRef
9. Zhang RH, Guo HY, Deng H, Li J, Quan ZhSh. Piperazine skeleton in the structural modification of natural products: a review. J Enzyme Inhib Med Chem. 2021;36(1):1165–97. doi: CrossRef
10. Romanelli MN, Manetti D, Braconi L, Dei S, Gabellini A, Teodori E. The piperazine scaffold for novel drug discovery efforts: the evidence to date. Expert Opin Drug Discov. 2022;17(9):969–84. doi: CrossRef
11. Vitaku E, Smith DT, Njardarson JT. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J Med Chem. 2014;57(24):10257–71024. doi: CrossRef
12. Brito AF, Moreira LKS, Menegatti R, Costa EA. Piperazine derivatives with central pharmacological activity used as therapeutic tools. Fundam Clin Pharmacol. 2019;33(1):13–24. doi: CrossRef
13. Hetzer HB, Robinson RA, Bates RG. Dissociation constants of piperazinium ion and related thermodynamic quantities from 0 to 50.deg. J Phys Chem. 1968;72(6):2081–6. doi: CrossRef
14. Manallack DT. The pKa distribution of drugs: application to drug discovery. Perspect Medicin Chem. 2007;1:25–38.
15. Viegas-Junior C, Danuello A, da Silva Bolzani V, Barreiro EJ, Fraga CA. Molecular hybridization: a useful tool in the design of new drug prototypes. Curr Med Chem. 2007;14(17):1829–52. doi: CrossRef
16. Szumilak M, Wiktorowska-Owczarek A, Stanczak A. Hybrid drugs-A strategy for overcoming anticancer drug resistance? Molecules. 2021;26(9):2601. doi: CrossRef
17. Rodríguez-Franco MI, Fernández-Bachiller MI, Pérez C, Hernández-Ledesma B, Bartolomé B. Novel tacrine-melatonin hybrids as dual-acting drugs for Alzheimer disease, with improved acetylcholinesterase inhibitory and antioxidant properties. J Med Chem. 2006;49(2):459–62. doi: CrossRef
18. Li K, Schurig-Briccio LA, Feng X, Upadhyay A, Pujari V, Lechartier B, et al. Multitarget drug discovery for tuberculosis and other infectious diseases. J Med Chem. 2014;57(7):3126–39. doi: CrossRef
19. Ciba Geigy Corp, Southcott MR. Novel oligomers useful for making cured fibre reinforced composites, US patent, US5026871A; 1991.
20. Kami?ski K, Obniska J, Wiklik B, Atamanyuk D. Synthesis and anticonvulsant properties of new acetamide derivatives of phthalimide, and its saturated cyclohexane and norbornene analogs. Eur J Med Chem. 2011;46(9):4634–41. doi: CrossRef
21. Sakhautdinov IM, Mukhametyanova AF. Synthesis of New cyclopentenofullerenes containing a norbornene fragment. Russ J Org Chem. 2019;55(9):1275–9. doi: CrossRef
22. Abagyan R, Totrov M, Kuznetsov D. ICM – A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. J Comput Chem. 1994;15(5):488–506. doi: CrossRef
23. Landrum, G. Rdkit: open-source cheminformatics software. [cited 2023 Apr 29] Available from: https://github.com/rdkit
24. Kumar PR, Seshadri M, Jaikrishan G, Das B. Effect of chronic low dose natural radiation in human peripheral blood mononuclear cells: evaluation of DNA damage and repair using the alkaline comet assay. Mut Res. 2015;775:59–65. doi: CrossRef
25. Dai C, Ellisen LW. Revisiting androgen receptor signaling in breast cancer. Oncologist. 2023;28(5):383–91. doi: CrossRef
26. Ravaioli S, Maltoni R, Pasculli B, Parrella P, Giudetti AM, Vergara D, et al. Androgen receptor in breast cancer: the “5W” questions. Front Endocrinol. 2022;13:977331. doi: CrossRef
27. Brisken C. Progesterone signalling in breast cancer: a neglected hormone coming into the limelight. Nat Rev Cancer. 2013:13(6):385–96. doi: CrossRef
28. Lydon JP, DeMayo FJ, Funk CR, Mani SK, Hughes AR, Montgomery CA Jr, et al. Mice lacking progesterone receptor exhibit pleiotropic reproductive abnormalities. Genes Dev. 1995;9(18):2266–78. doi: CrossRef
29. Zheng N, Chen J, Liu W, Liu J, Li T, Chen H, et al. Mifepristone inhibits ovarian cancer metastasis by intervening in SDF-1/CXCR4 chemokine axis. Oncotarget. 2017;8(35):59123–35. doi: CrossRef
30. Lee O, Sullivan ME, Xu Y, Rogers C, Muzzio M, Helenowski I, et al. Selective progesterone receptor modulators in early-stage breast cancer: a randomized, placebo-controlled phase II window-of-opportunity trial using telapristone acetate. Clin Cancer Res. 2020;26(1):25–34. doi: CrossRef
31. Liu JH, Soper D, Lukes A, Gee P, Kimble T, Kroll R, et al. Ulipristal acetate for treatment of uterine leiomyomas a randomized controlled trial. Obstet Gynecol. 2018;132(5):1241–51. doi: CrossRef
32. Lewis JH, Cottu PH, Lehr M, Dick E, Shearer T, Rencher W, et al. Onapristone extended release: safety evaluation from phase I–II studies with an emphasis on hepatotoxicity. Drug Saf. 2020;43:1045–55. doi: CrossRef
33. Ciebiera M, Vitale SG, Ferrero S, Vilos GA, Barra F, Caruso S, et al. Vilaprisan, a new selective progesterone receptor modulator in uterine fibroid pharmacotherapy-will it really be a breakthrough?. Curr Pharm Des. 2020;26(3):300–9. doi: CrossRef
34. West DC, Pan D, Tonsing-Carter EY, Hernandez KM, Pierce CF, Styke SC, et al. GR and ER coactivation alters the expression of differentiation genes and associates with improved ER+ breast cancer outcome. Mol Cancer Res. 2016;14(8):707–19. doi: CrossRef
35. Abduljabbar R, Negm OH, Lai CF, Jerjees DA, Al-Kaabi M, Hamed MR, et al. Clinical and biological significance of glucocorticoid receptor (GR) expression in breast cancer. Breast Cancer Res Treat. 2015;150(2):335–46. doi: CrossRef
36. Pan D, Kocherginsky M, Conzen SD. Activation of the glucocorticoid receptor is associated with poor prognosis in estrogen receptor-negative breast cancer. Cancer Res. 2011;71(20):6360-70. doi: CrossRef
37. Mitre-Aguilar IB, Moreno-Mitre D, Melendez-Zajgla J, Maldonado V, Jacobo-Herrera NJ, Ramirez-Gonzalez V, et al. The role of glucocorticoids in breast cancer therapy. Curr Oncol. 2022;30(1):298–314. doi: CrossRef
38. Baell JB, Nissink J, Willem M. Seven year itch: pan-assay interference compounds (PAINS) in 2017—utility and limitations. ACS Chem Biol. 2018;13(1):36–44. doi: CrossRef
39. Pham-The H, Cabrera-Pérez MÁ, Nam NH, Castillo-Garit JA, Rasulev B, Le-Thi-Thu H, et al. In silico assessment of ADME properties: advances in Caco-2 cell monolayer permeability modeling. Curr Top Med Chem. 2018;18(26):2209–29. doi: CrossRef
40. Wishart DS, Knox C, Guo AC, Cheng D, Shrivastava S, Tzur D, et al. DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008;36:D901–6. doi: CrossRef
41. Rampe D, Brown AM. A history of the role of the hERG channel in cardiac risk assessment. J Pharmacol Toxicol Methods. 2013;68(1):13–22. doi: CrossRef
42. Lionetto MG, Caricato R, Calisi A, Giordano ME, Schettino T. Acetylcholinesterase as a biomarker in environmental and occupational medicine: new insights and future perspectives. BioMed Res Int. 2013;2013:321213. doi: CrossRef
43. Elliott S. Current awareness of piperazines: pharmacology and toxicology. Drug Test Anal. 2011;3(7–8):430–8. doi: CrossRef
44. Zhou ZX, Yin XD, Zhang Y, Shao QH, Mao XY, Hu WJ, et al. Antifungal drugs and drug-induced liver injury: a real-world study leveraging the FDA adverse event reporting system database. Front Pharmacol. 2022;13:891336. doi: CrossRef
45. Bunchorntavakul Ch, Reddy KR. Drug hepatotoxicity: newer agents. Clin Liver Dis. 2017;21(1):115–34. doi: CrossRef
46. Yarlagadda SG, Perazella MA. Drug-induced crystal nephropathy: an update. Expert Opin Drug Saf. 2008;7(2):147–58. doi: CrossRef
47. Ballantyne B, Myers RC, Klonne DR. Comparative acute toxicity and primary irritancy of the ethylidene and vinyl isomers of norbornene. J Appl Toxicol. 1997;17(4):211-221. doi: CrossRef
48. Rheingold SR, Neugut AI, Meadows AT. Therapy-related secondary cancers. 6th ed. BC Decker: Holland-Frei Cancer Medicine; 2003.
49. ICH S1A. Guideline on the need for carcinogenicity studies of pharmaceuticals. S1A Guideline. 1995. [cited 2024 Sep 9]. Available from https://www.ich.org/page/safety-guidelines
50. ICH topic S1B. Carcinogenicity: testing for carcinogenicity of pharmaceuticals. Step 4 consensus guideline. Part I. S1B-R1. 2022. [cited 2024 Sep 10]. Available from https://www.ich.org/page/safety-guidelines
51. ICH topic S1B. Testing for carcinogenicity of pharmaceuticals, part II, S1B-R1. 2021. [cited 2024 Sep 10] Available from https://www.ich.org/page/safety-guidelines
52. Krewski D, Acosta D Jr, Andersen M, Anderson H, Bailar JC 3rd, Boekelheide K, et al. Toxicity testing in the 21st century: a vision and a strategy. J Toxicol Environ Health B Crit Rev. 2010;13(2–4):51–138. doi: CrossRef
53. Horwitz KB, Costlow ME, McGuire WL. MCF-7; a human breast cancer cell line with estrogen, androgen, progesterone, and glucocorticoid receptors Steroids. 1975;26(6):785–95. doi: CrossRef
54. Choupani E, Madjd Z, Saraygord-Afshari N, Kiani J, Hosseini A. Combination of androgen receptor inhibitor enzalutamide with the CDK4/6 inhibitor ribociclib in triple negative breast cancer cells. PLoS One. 2022;17(12):e0279522. doi: CrossRef
55. Lee YHA, Hui JMH, Leung CH, Tsang CTW, Hui K, Tang P, et al. Major adverse cardiovascular events of enzalutamide versus abiraterone in prostate cancer: a retrospective cohort study. Prostate Cancer Prostatic Dis. 2023;27(4):776–82. doi: CrossRef
56. Strehl C, Ehlers L, Gaber T, Buttgereit F. Glucocorticoids-all-rounders tackling the versatile players of the immune system. Front Immunol. 2019;10:1744. doi: CrossRef
57. Kadmiel M, Cidlowski JA. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol Sci. 2013;34(9):518–30. doi: CrossRef
58. Buxant F, Kindt N, Laurent G, Noël JC, Saussez S. Antiproliferative effect of dexamethasone in the MCF-7 breast cancer cell line. Mol Med Rep. 2015;12(3):4051–4. doi: CrossRef
59. Serbian I, Hoenke S, Csuk R. Synthesis of some steroidal mitocans of nanomolar cytotoxicity acting by apoptosis. Eur J Med Chem. 2020;199:112425. doi: CrossRef
60. Tieszen CR, Goyeneche AA, Brandhagen BN, Ortbahn CT, Telleria CM. Antiprogestin mifepristone inhibits the growth of cancer cells of reproductive and non-reproductive origin regardless of progesterone receptor expression. BMC Cancer. 2011;11:207. doi: CrossRef
61. Araldi RP, de Melo TC, Mendes TB, de Sá Júnior PL, Nozima BH, Ito ET, et al. Using the comet and micronucleus assays for genotoxicity studies: a review. Biomed Pharmacother. 2015;72:74–82. doi: CrossRef
62. Azqueta A, Stopper H, Zegura B, Dusinska M, Møller P. Do cytotoxicity and cell death cause false positive results in the in vitro comet assay?. Mutation Res Genet Toxicol Environ Mutagen. 2022;881:503520. doi: CrossRef
63. Sakellakis M, Flores LJ. Androgen receptor signaling–mitochondrial DNA–oxidative phosphorylation: a critical triangle in early prostate cancer. Curr Urol. 2022;16(4):207–12. doi: CrossRef
SUPPLEMENTARY MATERIAL
The supplementary material can be accessed at the link here: [https://japsonline.com/admin/php/uploadss/4540_pdf.pdf]