
INTRODUCTION
As per the reports shared by GLOBOCAN in 2022, the number of new cases and deaths from Cancer was estimated to be 20 and 9.7 million, respectively [1]. The main cause attributed to carcinogenesis is the disrupted gene regulation, which may be potentiated by various risk factors leading to increased prevalence of disease in patients across all age groups [2]. The burden of cancer is expected to be raised to 28.4 million by 2040, which is an estimated increase of 47% when compared against 2022 [3]. The main contributing factors for this significant increase in the prevalence of cancer are attributed to various exogenous (e.g., smoking and lifestyle changes), extrinsic (e.g., immune, microbiota, and hormonal), and intrinsic (e.g., DNA-related alterations) risk factors [4,5]. While these numbers are disturbing, astonishing, and difficult to digest, this also clearly indicates the inability of the currently available medications and the necessity to have more sophisticated, targeted therapies. Availability of novel therapies helps to target the tumor-specific sites thereby potentiating the efficacy of the treatment. These targeted therapies have proven their ability to enhance efficacy in various types of cancers including breast, colon, and small-cell lung cancers [6–8].
Among the targeted therapies, tyrosine kinase inhibitors (TKIs) have gained popularity as major treatment options in drug discovery. TKIs inhibit kinases from phosphorylating tyrosine residues of their substrates and eventually block the activation of downstream signaling pathways and disrupt the signaling pathways to prevent abnormal proliferation [9]. These agents have emerged as powerful treatment options for patients suffering from various cancers such as renal, hepatocellular carcinoma, and lung cancer [10–12]. Over the past 20 years, there have been more than 100 TKIs have been approved, targeting more than 58 receptor tyrosine kinases and 32 non-receptor kinases thereby enabling molecular targeting mechanisms [13,14]. In a recent review, Kollipara et al. [13] indicated that there are more than 70 TKIs have been approved by the United States Food and Drug Administration from 2021 for targeting EGFR, ALK, ROS1, HER2, and NTRK through molecular mechanisms, thereby portraying the quantum of research ongoing in this direction.
To exert any efficacy, it is essential that the drug formulated as a drug product should exert suitable absorption, distribution, metabolism, and elimination (ADME) to have clinically relevant concentrations at the site of action [15]. Together with the physicochemical properties of the drug, the pharmacokinetic variability also contributes to the drug availability at the target site [16]. In the case of TKIs, the dose as well as dosage regimen is appropriately selected by the oncologists with the aim to have the highest drug concentration at the target site for maximizing efficacy. Additional strategies such as dose reduction, dose escalation, and optimization to balance efficacy and adverse events are routinely followed by medical oncologists [17]. On the other hand, the highest doses of TKIs may be required to have a broad inhibitory profile and enduring clinical benefit [18]. Specific to TKIs, although they are highly efficacious and target specific tumor sites, their administration is associated with significant challenges related to food intake, drug–drug interactions (DDIs) [19]. TKIs exhibit pH-dependent solubility and because of their weakly basic nature, while they get solubilized in the stomach, they may be subjected to precipitation upon entry to the intestine due to increased pH. Because of these aspects, many TKIs including dasatinib, bosutinib, and gefitinib possess challenges for sub-optimal exposures [20]. This aspect becomes prominent as these drugs are used to treat cancers such as chronic myeloid leukemia (dasatinib, bosutinib) and non-small cell lung carcinoma (gefitinib). The situation of sub-optimal exposures gets much more pronounced when TKIs are co-administered with acid reducing agents (ARAs) such as proton pump inhibitors (PPI), H2 receptor antagonists (H2RA), or antacids. As the administration of ARA increases the stomach pH, the solubility and dissolution rate of TKIs gets reduced because of precipitation at higher pH thereby leading to DDI. From the treatment perspective, ARAs are prescribed routinely in cancer patients to reduce gastric-related adverse events as well as to prevent gastric mucosal damage caused by cytotoxic chemotherapy [21,22]. However, because of the absorption level DDI between ARA and TKI, the exposure of the TKI gets reduced significantly thereby leading to a decreased survival rate and increased risk of death [23]. A typical practice of reducing the risk of such DDI is through appropriate label recommendation wherein staggering or complete avoidance of ARA may be suggested based on the level of interaction. However, if the recommendation of ARA is inevitable to enhance patient compliance and to reduce gastrointestinal side effects, then the efficacy of the TKI treatment is compromised. Thus, an ideal situation would be a scenario where DDI is mitigated to a greater extent through appropriate formulation innovation. Any formulation approach, that is leading to enhanced solubility of TKIs in the pH range of 4–6.8 may reduce the risk of precipitation thereby providing the possibility to overcome the absorption DDI between TKI and ARA. The literature review indicated the use of novel active pharmaceutical ingredient (API) based or formulation-based interventions that circumvented absorption level DDIs [23,24]. However, such formulation optimizations are time and resource consuming and often require significant efforts to come up with suitable formulation and subsequent clinical studies to demonstrate the absence of DDIs. To reduce the effort towards formulation development, optimization, and clinical evaluations, modeling approaches such as PBBM modeling can be utilized effectively for rational formulation development. Modeling and simulation approaches such as PBBM have gained popularity in recent years due to their plethora of applications at both new drug and generic product development. These models are used to select appropriate formulations for first-in-human studies, evaluating DDIs, establishing dissolution specifications, and enabling biowaivers [25–31]. In the present work, we have successfully utilized PBBM to model and predict, and mitigate the DDIs arising from interactions of TKIs and ARA.
Three drugs were selected for the current study, namely dasatinib, bosutinib, and gefitinib for which ARA restriction has been included in the label due to significant DDIs (Table 1). For all three drugs, using pH versus solubility, permeability, physiology, and pharmacokinetic inputs, PBBM was developed in the fasting condition. The developed models were validated across different oral and intravenous doses and, subsequently, DDI potential was predicted by increasing the stomach pH to 5, mimicking PPI administration. The observed ARA interaction effect was compared against that of clinically reported for determining model credibility. Subsequently, the pH versus solubility data generated in the presence of citric acid was inputted in the model and the DDI potential was estimated. The predicted DDI potential with citric acid-based solubility was compared against that of without citric acid to determine reduced DDI potential due to increased solubility in the pH range of 4–6.8. Subsequently, sensitivity of model towards precipitation potential for all drugs was determined in normal stomach condition (pH 1.3) as well as in altered stomach condition (pH 5) mimicking PPI administration. Further, due to enhanced solubility in the presence of citric acid, possibility of dose reduction has been evaluated for selected drugs. The physical situation being modeled in the present study is to mimic administration of TKIs and ARAs together. Due to significant DDI, such an administration scheme is not possible and thus label has mentioned the restrictions. However, with the help of suitable formulation intervention, such DDIs can be avoided. Thus, our study aimed to mimic a physical situation where TKI and ARA drugs are taken together. Overall, this work portrays the importance of formulation intervention to mitigate ARA impact of TKIs and the utility of PBBM to support the successful formulation development of TKIs to enhance the efficacy of treatment.
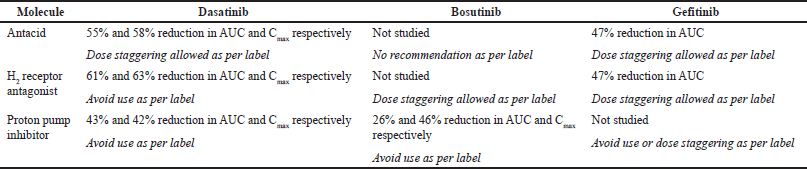 | Table 1. Literature reported ARAs impact on selected drugs. [Click here to view] |
MATERIALS AND METHODS
Materials
The APIs dasatinib, bosutinib, and gefitinib were obtained as gift samples from Hetero Drugs, Hyderabad. All the reagents used in this study for solubility determination and analytical methodology were of either analytical or HPLC grade. Deionized distilled water was obtained from a Milli-Q water purification system (Millipore, MA). For the solubility study, all the aqueous buffers (pH 2, 3, 4.5, 5, and 6.8) were prepared based on United States Pharmacopoeia [32]. Physicochemical and biopharmaceutical parameters used for the development of PBBM for dasatinib, bosutinib, and gefitinib were primarily collected from available literature, except for the solubility which is generated experimentally. In the absence of literature or in-house data, ADMET-predicted values were utilized for model development. Gastroplus, version 9.9.002 (Simulations Plus Inc., Lancaster, California), was used for the development and validation of PBBM (referred as Gastroplus in later parts of the manuscript). Several Gastroplus modules, i.e., Advanced Compartmental Absorption and Transit (ACATTM), ADMET® Predictor, PKPlusTM, and OptimizationTM tool were used during model development.
Solubility study
The solubility study was performed using the shake flask method. Aqueous buffers (plain) and aqueous buffers containing 0.2 mg/ml citric acid (5 ml each) were placed in glass vials and kept in a heating orbital shaker at 37°C and 100 rpm for 15–20 minutes to allow the buffers to reach the desired temperature. Considering the average dosage form weight of 500 mg tablet, the amount of acidifier (i.e., citric acid) was considered at 10%, equivalent to 50 mg. Considering the stomach volume of 250 ml, citric acid concentration in the stomach may result in 0.2 mg/ml, and thus, the same concentration was chosen for the solubility experiment. During solubility determination, a weighed amount of the API (dasatinib, bosutinib, and gefitinib) was then added to each vial to create a saturated solution. Samples (150 μl) were withdrawn at different time points (2, 4, and 6 hours), centrifuged for 5 minutes at 12,000 rpm, and the supernatant was analyzed using HPLC after appropriate and immediate dilution to determine the amount of drug solubilized. The solubility data was determined in triplicates and mean, standard deviation were reported for both solubility determinations (with and without citric acid). For the solubility study, a heating orbital shaking incubator (REMI, RIS-24 Plus) and centrifuge (Eppendorf, 5430 R) were used.
HPLC analysis
Chromatographic analysis of the samples obtained from the solubility study was performed using a Shimadzu Prominence HPLC system (Shimadzu Corporation, Kyoto, Japan). The system was equipped with a degasser unit (DGU-20A3R), dual pumps (LC-20AD), an autosampler (SIL-20ACHT), a column oven (CTO-20AC) with temperature control, and a photodiode array-UV detector (SPD-M20A). Data obtained from the HPLC system were analyzed using LC Solutions software (version 1.25, Shimadzu Corporation). All chromatographic separations were carried out on RP18 column (125 × 4 mm, 5 μm) with a flow rate of 1 ml/min. Mobile phase employed was 20 mM Ammonium acetate buffer pH 6.6 and Acetonitrile at 65:35 (%v/v) (dasatinib), 20 mM Ammonium acetate buffer pH 4 and Acetonitrile at 65:35 (%v/v) ratio (bosutinib) and 20 mM Ammonium formate buffer pH 3.5 and Acetonitrile at 75:25 (%v/v) ratio (gefitinib). Separations were carried out at column oven temperatures of 25°C, 45°C, and 40°C were employed for dasatinib, bosutinib, and gefitinib. An injection volume of 10–20 μl was employed during the analysis and wavelengths of 310, 260, and 248 nm were used for dasatinib, bosutinib, and gefitinib, respectively. Using these chromatographic techniques, a retention time between 4 and 6 minutes was obtained for the selected molecules.
In vivo data for PBBM
The in vivo data required for PBBM development and validation for all three molecules is gathered from the literature. Dasatinib oral pharmacokinetic profiles for 70 and 100mg doses were gathered from Kovar et al. [33]. Bosutinib intravenous infusion (120 mg, 1 hour), and oral pharmacokinetics data for 200, 400, and 500 mg were obtained from the literature [34–37]. Gefitinib intravenous infusion (100 mg, 1 hour), and oral pharmacokinetic data of 250 mg were used from the literature [38]. All the pharmacokinetic profiles (time, mean, and standard deviation) were digitized for the purpose of PBBM development and validation.
PBBM modeling and simulation approach
Gastroplus was used to build the PBBM in fasting conditions for dasatinib, gefitinib, and bosutinib. Human gastrointestinal physiology was incorporated into the model through ACATTM. This model accounted for all the processes namely solubilization, dissolution, precipitation, permeation, metabolism, and presence in systemic circulation for all the selected drugs. Additional features such as ADMET PredictorTM and PKPlusTM were used to obtain in silico estimates of model parameters and to determine the elimination parameters of the model. The workflow used for the modeling and simulation exercise in the present work is described in Figure 1. The parameters used for model development are described in the following sections.
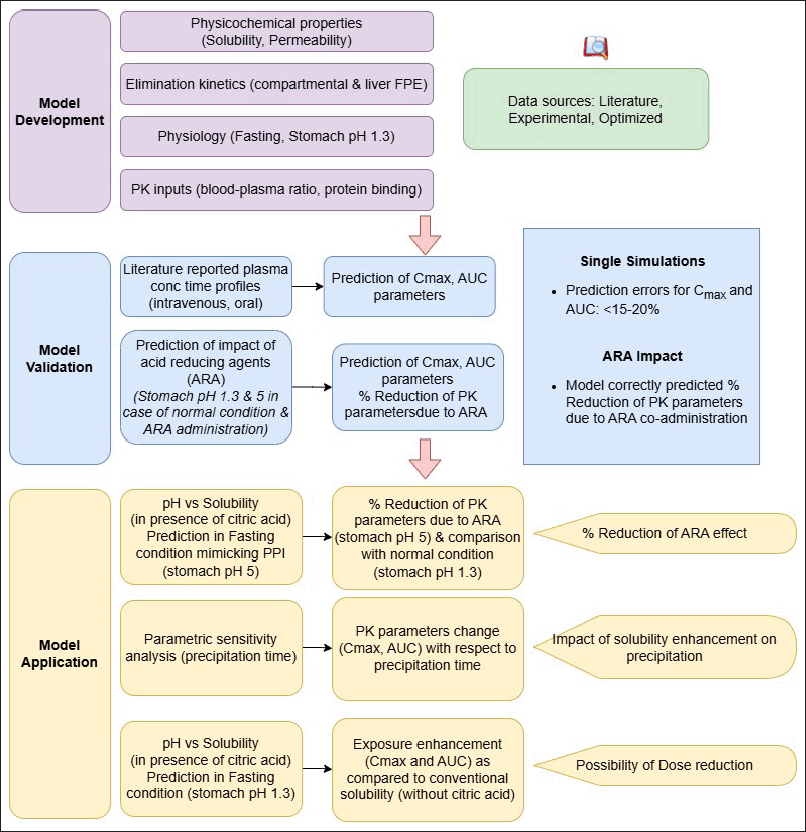 | Figure 1. Gastroplus modeling workflow for evaluation of ARA interactions for selected drugs (dasatinib, bosutinib, gefitinib). [Click here to view] |
Physicochemical and biopharmaceutics properties
The physicochemical and biopharmaceutical properties used for the PBBM development for all three molecules dasatinib, bosutinib, and gefitinib are provided in Table 2. The solubility input used in the model (i.e., pH vs. solubility) with and without citric acid is provided in Table 3. For initial model development, pH versus solubility profile of all the drugs without citric acid is used (termed as conventional solubility). For all the simulations, immediate release tablet was selected as dosage form. For the fasting simulations, 250 ml of water is consumed and same volume as the administration volume is considered during all the simulations. Particle size (i.e., diameter) of 25 μm was considered during all simulations as a default value. During simulations, the in vivo dissolution is determined based on particle size, diffusion layer thickness, effective surface area, and diffusion coefficient. Furthermore, a default precipitation time of 900 seconds was used for both dasatinib and gefitinib whereas for bosutinib this value is optimized to 2,000 seconds due to low probability of precipitation at intestinal condition based on solubility data presented in Table 3.
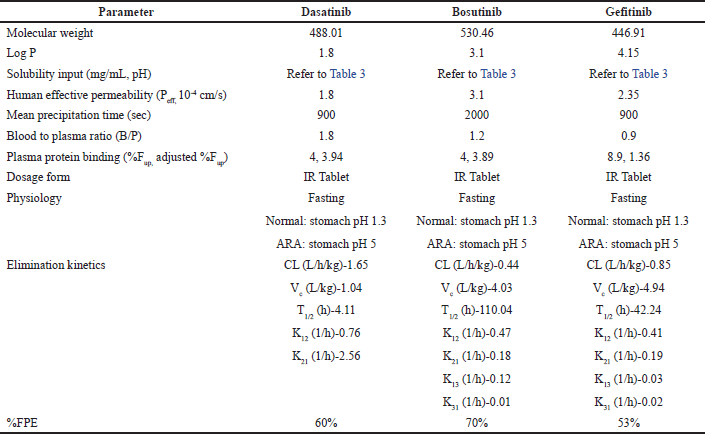 | Table 2. Gastroplus model inputs for selected drugs. [Click here to view] |
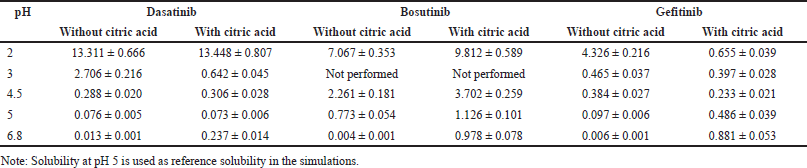 | Table 3. pH versus solubility data of selected drugs (mean ± SD, n = 3). [Click here to view] |
Physiology
For the simulation of in vivo pharmacokinetic study in the fasting condition for all three drugs, “Human physiology-fasting” was considered. The absorption scaling factor model used in the present simulations is Opt Log D Model SA/V 6.1. The developed oral mechanistic model accounted for passive absorption and no active transport is included. For all three drugs, ADMET predictor predicted permeability was used during initial simulations and was later optimized to account for in vivo Tmax accurately for oral pharmacokinetic data. The optimized permeability values for all three drugs are presented in Table 2.
Elimination parameters
For physiological models, the best way to describe the elimination parameters is based on intravenous pharmacokinetic data [39,40]. However, in the absence of the intravenous pharmacokinetic data, the elimination parameters can be determined after correcting for bioavailability. In the case of dasatinib, the intravenous pharmacokinetic data is not available, however in preclinical species, the bioavailability ranged from 14% to 34% [41]. Considering the average bioavailability of 20% for humans, the elimination parameters were determined for 100 mg oral profile using 2-compartmental model and the bioavailability was corrected using the liver first pass extraction (%FPE) as indicated in Table 2. In the case of bosutinib and gefitinib, due to the intravenous pharmacokinetic data availability, the oral as well as intravenous pharmacokinetic data fitted together, and elimination parameters, %FPE were determined. In both cases, the elimination parameters followed 3-compartmental model and are presented in Table 2. Other pharmacokinetic inputs such as protein biding and blood-to-plasma ratio are determined from the literature and is indicated in Table 2. Further, a default body weight of 70 kg was used during all simulations for adult healthy human physiology.
Model validation
The model was developed using the above-mentioned model input parameters and was validated using the literature-reported plasma concentration profiles of intravenous infusion as well as oral formulations. For the model validation exercise, prediction error (%PE) as well as folds error (FE) were calculated using the formulas (1) and (2). Both parameters were used to obtain confidence into the developed PBBM for all three drugs [42,43].
Further, the model’s ability to correctly predict the ARA effect reported in the literature (Table 3) is evaluated [44–46]. In the case of ARA administration, on average, the stomach pH gets increased to 5 and this change was made to simulate ARA administration [47]. Further, the %reduction of AUC parameter in the case of ARA administration is calculated using the formula (3) and this predicted value is compared against observed data in the literature to demonstrate models’ credibility for all three drugs. For this purpose, 100, 400, and 250 mg oral doses were used for dasatinib, bosutinib, and gefitinib, respectively [47].
Model application
The validated PBBM for all three drugs were subsequently applied with an objective to evaluate the ARA and TKI DDI mitigation strategies as follows.
DDI mitigation due to acidifying agent
The addition of citric acid in the formulation helps to provide acidifying microenvironment in the formulation to enhance the solubility of TKI in the region of pH 4.5–6.8. Further, to evaluate the impact of citric acid on DDI mitigation, the solubility data generated in the presence of citric acid (Table 3) is inputted into the model and the AUC values were simulated at normal stomach pH condition (pH 1.3), and pH condition mimicking ARA administration (pH 5). The % reduction in AUC with this set of solubility data is determined using formula (3).
Solubility enhancement on precipitation behavior
Due to solubility enhancement, especially in the pH range of 4–6.8, there can be the possibility of reduced precipitation of the TKIs upon entering the intestine, thereby, avoiding such potential limitation for absorption. For evaluating this behavior, the in vivo dissolution profiles of all three drugs were determined during the simulations. The in vivo dissolution profiles were compared with conventional and enhanced solubility (with citric acid) under normal stomach (pH 1.3) and the stomach condition (pH 5) mimicking ARA administration. Further, parametric sensitivity analysis (PSA) analysis was performed under a normal stomach (pH 1.3) with respect to precipitation time for evaluation of the impact of solubility on precipitation.
Possible reduction in the dose
Due to solubility enhancement in the presence of citric acid, there might be possible dose reduction as the increased solubility can enhance exposures. Thus, the possibility of dose reduction and the folds enhancement in exposures are calculated for all three drugs using the formulas (4) and (5) by performing the simulations in fasting stomach condition (pH 1.3).
RESULTS
In the present study, PBBM was developed to predict and to demonstrate the mitigation of ARA, and TKIs DDI by using acidifying agent in the formulation (Fig. 1). All PBBMs were developed through the integration of physicochemical, pharmacokinetic, and physiological properties. The developed models enabled multiple applications with respect to the mitigation of DDI, possible dose reductions, and lowered precipitation potential.
Solubility
The solubility study was conducted at 37°C across the pH conditions of 2–6.8 with and without citric acid (at a concentration of 0.2 mg/ml). The generated solubility data is presented in Table 3 and for all the drugs, pH-dependent solubility has been observed wherein solubility was found to be higher in acidic conditions followed by reduced solubility as the pH increased. The data of three replicates was found to be closer to each other and the variability was found to be lower thereby demonstrating the reliability of the results. When compared with conventional solubility (without citric acid), the presence of citric acid enhanced the solubility especially at the pH conditions of 4.5–6.8 thereby confirming that an acidic environment facilitated more solubilization. This phenomenon was observed for all the studied drugs and specifically, significant improvement in solubility is seen at pH 6.8 due to the presence of citric acid.
PBBM simulations
Model development
Initial PBBM was developed for all the drugs, dasatinib, bosutinib, and gefitinib using the physicochemical properties, pharmacokinetic, and physiological parameters listed in Table 2 and using conventional solubility in Table 3. The elimination kinetics calculated either from the intravenous and oral data (bosutinib, gefitinib) or solely from the oral data (dasatinib) depicted the plasma concentration values appropriately. Further, optimization of precipitation time in the case of bosutinib also ensured matching predictions with that of clinical data thereby enabling model suitability. The incorporation of %FPE to enable accurate bioavailability consideration in the model has yielded acceptable PBBMs for all the molecules. Further, optimized permeability resulted in predictions that are matching with literature-reported data. Human physiology fasting has demonstrated its suitability in the current simulations and thus didn’t require any modifications. Overall, together with all model inputs, the models demonstrated acceptability for all the molecules studied.
Model validation
After successful model development, detailed validation has been performed across the dosage forms and doses for all the selected drugs. For dasatinib, model validation has been performed with oral doses whereas for bosutinib and gefitinib, model validation is performed using both intravenous infusion and oral doses. The results of the model validation are depicted in Table 4, Figures 2 and 3. As indicated in Figure 2, for all tree drugs, there is a good agreement between observed versus predicted plasma concentration-time data. The predictions are closer to the observed data and were within the variability observed. As can be seen from Table 4 that the PEs for Cmax and AUC were acceptable across different dosage forms and different doses thereby confirming the validity of the developed models. Apart from PEs, the FEs were also closer to 1 thereby confirming the validity of the model. Further, it is evident from Figure 3 that both the predicted Cmax and AUC values within acceptable folds limits as that of the observed data. In most of the cases, the observed versus predicted values are closer to unity and almost all the cases, the results were within 1.25 folds thereby confirming the validity of the developed PBBM. Further, the predictions across dosage forms and doses provided stronger confidence in the models, and subsequently, the models were used to predict the literature reported ARA effect.
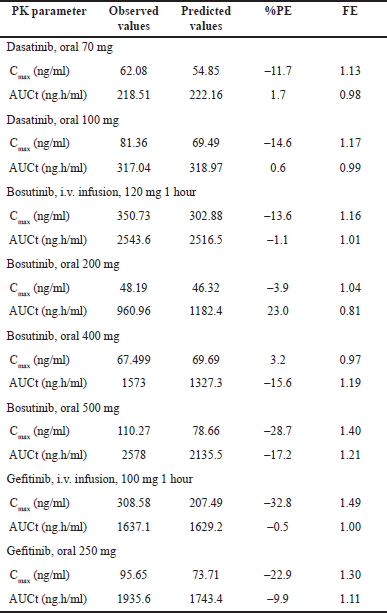 | Table 4. Model validation for selected drugs (observed vs. predicted) %PE, FE. [Click here to view] |
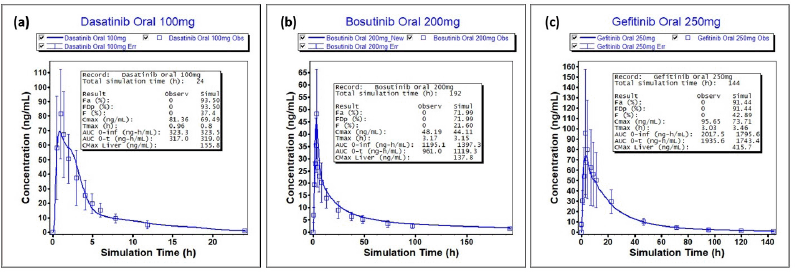 | Figure 2. Gastroplus model validation against oral data (a) dasatinib oral 100mg (b) bosutinib oral 200 mg (c) gefitinib oral 250 mg. Horizontal axis: time and vertical axis: plasma concentration. [Click here to view |
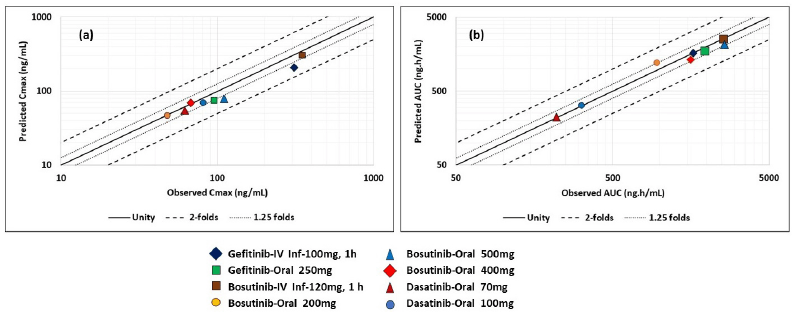 | Figure 3. Predictability graphs for Cmax and AUC parameters across the doses and dosage forms for selected drugs (a) Cmax (b) AUCt. Horizontal axis: observed parameter and vertical axis: predicted parameter. [Click here to view] |
To simulate the ARA effect, the predictions for AUC have been performed with a normal stomach (pH 1.3) and an altered stomach (pH 5) due to PPI administration. The AUC values were simulated in both cases and the ratio was determined and subsequently %AUC reduction was calculated. The comparison observed versus simulated ARA DDI is depicted in Figure 4 and represented in Table 5. The simulation results indicated that all the models predicted the ARA effect accurately in comparison with literature-reported data. The data from Figure 4 indicated that the observed versus predicted DDI is within 1.25 folds for two cases (dasatinib, gefitinib) and within 2 folds for one case (bosutinib). The observed DDI effect for bosutinib is significantly lesser (17%) as compared to other drugs and thus the predictions within two folds for this molecule were considered to be acceptable. Overall, the developed PBBM simulated DDI effect well for all the studied drugs.
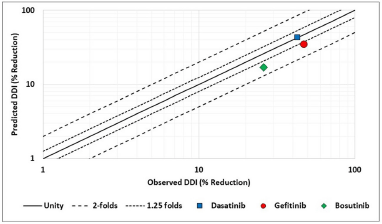 | Figure 4. Predictability graphs for observed versus predicted ARA interaction. Horizontal axis: observed interaction and vertical axis: predicted interaction. [Click here to view] |
 | Table 5. Observed versus predicted PPI effect and simulated PPI effect with citric acid. [Click here to view] |
Model application
The fully validated PBBM was successfully used for various applications as mentioned below.
DDI mitigation due to acidifying agent
To evaluate the impact of citric acid addition on the DDI mitigation, the model predictions were performed with solubility generated in the presence of citric acid at both normal stomach (pH 1.3) as well as at stomach (pH 5) mimicking ARA administration. The DDI effect in this scenario was calculated and presented in Table 5 and Figure 5 in comparison against that of simulated DDI effect without citric acid addition. In conjunction with the increased solubility in the presence of citric acid (Table 3), the results demonstrated that the DDI effect is almost nullified due to the presence of citric acid. As it can be seen from Table 5 the % reduction in AUC is almost close to zero thereby indicating the possibility of complete DDI mitigation. Overall, from Figure 5, it is evident that the predicted DDI effect is significantly reduced for all the studied molecules.
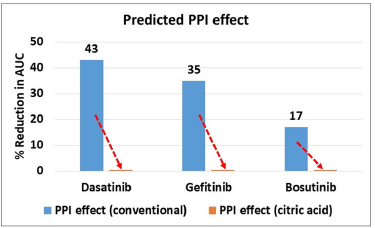 | Figure 5. Predicted PPI effect (with citric acid) against conventional (without citric acid) for selected drugs. [Click here to view] |
Solubility enhancement on precipitation behavior
To understand the impact of solubility enhancement on precipitation behavior, the in vivo dissolution curves were simulated at different conditions using conventional solubility and solubility in the presence of citric acid at normal stomach (pH 1.3) and stomach (pH 5) mimicking ARA administration. The result of this exercise is presented in Figure 6 wherein with conventional solubility, solubilization followed by precipitation is observed for all the molecules. With conventional solubility, when stomach pH is increased to 5, the in vivo dissolution was slower and incomplete to solubilize the entire dose of the drugs, thereby, demonstrating potential DDI. When solubility with citric acid is incorporated in the model, the models predicted faster in vivo dissolution without precipitation thereby indicating a positive impact of solubility. Further, the in vivo dissolutions were similar in the normal stomach (pH 1.3) and stomach (pH 5) mimicking ARA administration thereby indicating the absence of interaction. Additionally, no precipitation behavior was seen at both stomach pH of 1.3 and 5 when citric acid-based solubility was used. Further, PSA analysis performed in a normal stomach (pH 1.3) using conventional and citric acid solubility (Fig. 6) indicated that precipitation time has sensitivity towards AUC when conventional solubility is used whereas there is no impact of precipitation time on AUC when citric acid-based solubility was used. This aspect clearly demonstrates the nullified in vivo precipitation when solubility is enhanced in the range of pH 4.5–6.8 for all the drugs.
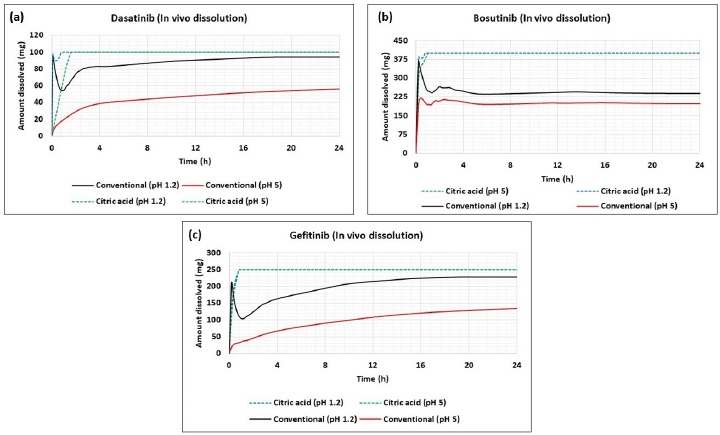 | Figure 6. In vivo dissolution graphs for selected drugs (a) dasatinib (b) bosutinib (c) gefitinib. Horizontal axis: time and vertical axis: amount dissolved (mg). [Click here to view] |
Possible reduction in the dose
Due to solubility enhancement in the presence of citric acid, possibility of dose reduction for all drugs has been studied and the results are portrayed in Table 6. When AUC values generated with citric acid solubility is compared against conventional solubility, a fold’s increase of 1.08 1.60, and 1.10 was observed for dasatinib, bosutinib, and gefitinib resulting in dose reduction possibility by 7%, 37.5% and 8.8% respectively, owing to enhancement in solubility. These values represent mean reduction in doses and the ranges, confidence intervals of the dose adjustments can be estimated in the future based on the population simulations as described in the discussion section. Further, the reduced doses are deemed to yield bioequivalence against innovator formulation and thus similar clinical response is anticipated yet providing flexibility in dose administration with ARAs.
 | Table 6. Dose reduction possibility with increased solubility in presence of citric acid. [Click here to view] |
DISCUSSION
TKIs have demonstrated significant efficacy against various types of cancers including breast cancer, colon cancer, lung cancer, and so on, due to molecular targeting mechanisms. Over the past two decades, more than 100 TKIs have been approved thereby portraying the extensive research done in medical oncology. Even though TKIs are efficient in various types of cancers through molecular targeting mechanisms, they are victims of administration challenges [13]. The TKIs are weakly basic in nature and thus are prone to precipitation in the intestinal condition, thereby leading to pharmacokinetic variability as they exhibit pH-dependent dissolution. Additionally, there has been inconsistent labeling practices observed for TKIs and despite positive food effects for many agents, these agents are administered in fasting conditions [48–50]. This aspect may be justifiable from a safety perspective, but administration in fasting conditions despite positive food effects may yield sub-optimal exposures leading to lower or insignificant therapeutic efficacy. Another important consideration in TKIs administration is the number of pills wherein some agents such as vemurafenib may require eight pills per day and neratinib require six pills per day [51,52]. Additionally, some of the agents require administration at multiple frequencies in a day (e.g., icotinib is administered three times a day) thereby possessing challenges to adhere to administration schemes.
Another important challenge in the administration of TKIs is the impact of ARAs on reduced exposures. Within ARA, there are further sub-classes of agents like antacids, H2RA, PPIs, and so on. Although all these agents reduce the exposure of TKIs due to increased stomach pH, PPIs represent the worst case due to their long-lasting effect of increased stomach pH [53,54]. The DDIs with PPIs may happen at the systemic level (due to inhibition or induction of metabolizing enzymes) or at the absorption level (due to increased stomach pH). In case the TKIs represent pH-dependent solubility, then the later type of interactions is prevalent. Administration of ARA during TKI treatment is necessary to avoid gastrointestinal issues and to prevent mucosal damage caused by cytotoxic treatment. Raoul et al. [55] studied the oncology drug records of approximately 872 and observed that more than one quarter of patients regularly use PPIs. Uchiyama et al. [56] reviewed the impact of PPI on the efficacy of oncology treatment and concluded that due to impaired efficacy, it was suggested to limit unnecessary over usage of PPIs. Lee et al. [57] studied the impact of PPI on the efficacy of palbociclib treatment in patients with breast cancer. It was concluded that overall survival in concomitant PPI group was shorter than that of the non-concomitant PPI group. These examples clearly indicate the clinically significant impact of ARA on the TKI treatment efficacy. While the PPIs cannot be completely avoided, alternatives to mitigate the DDI were studied in the literature. Buti et al. [58] indicated that refraining the PPIs could be an option but other approaches such as dose staggering can be evaluated to mitigate DDI. As indicated in Table 1, dose staggering is allowed in a few cases for all three drugs, depending on the extent of interaction observed and the type of ARA.
To further understand the mechanism of DDI of TKIs with ARAs, further investigation has been performed in the literature. Buddha et al. [59] studied the clinical literature of 15 TKIs exhibiting DDIs with ARA and concluded that the interaction ismaximum in cases where the solubility varies over the range of pH 1–4. As the TKIs are weakly basic in nature and exhibit pH-dependent solubility, the more the difference between solubility between pH 1 and 4, the more the possibility of interaction expected. Thus, the higher the steepness of the pH solubility curve, more is the interaction expected. Among the studied molecules in the present work, bosutinib has a lower steepness of the pH versus solubility curve and thus, a lower interaction of 17% reduction is seen for this molecule as compared to dasatinib and gefitinib (Table 3). Various approaches to increase the solubility in the range of pH 4.5–6.8 have been evaluated for TKIs to reduce the ARA impact and to relax the labeling schemes to allow staggering or co-administration. Hofmann et al. [24] portrayed the use of dasatinib anhydrate with improved solubility in the range of pH 4.5–6.8 as compared to monohydrate thereby enabling the staggered administration as compared to complete avoidance. Similarly, Larfors et al. [60] came up with a novel solid dispersion of dasatinib that has improved solubility throughout pH range thereby providing the possibility of co-administration of dasatinib with ARA. However, the development of such formulations requires multiple iterations and significant effort and thus, approaches such as PBBM offers attractive alternatives to extensive formulation experiments and clinical trials.
The use of PBBM to predict the DDI between TKI and ARA has been evaluated in the literature. Dodd et al. [47] use the modeling approach at the drug discovery and interface to demonstrate the utility of modeling approach to predict the DDI based on structure or based on minimum solubility data wherein a pH value of 5 is considered for simulations with ARAs. Mitra et al. [61] described the industry case studies wherein pH-dependent DDIs were evaluated by PBBM approach for weakly basic drugs. Based on the case studies described, a pragmatic PBBM workflow for the evaluation of such DDIs has been suggested. Although these case studies highlight the importance of PBBM in predicting DDIs between TKI and ARA accurately, PBBM utility to mitigate the risk of DDI using formulation intervention to proactively identify the mitigation was not discussed in detail. We could find only few publications wherein PBBM was used to predict pH-dependent DDI and one of such publication is Parrott et al. [62] wherein PBBM was used to explore gastric pH changes on the pharmacokinetics of entrectinib. It was indicated that the presence of acidifying agent in the formulation reduced the effect of gastric pH changes.
In the present manuscript, we attempted to demonstrate the use of PBBM not only to predict, but to mitigate the DDI risk through the addition of acidifying agent in the formulation to enhance solubility in the region of pH 4.5–6.8. The highlight and novelty of this present study is two folds: (1) To avoid DDI between TKI and ARA using suitable formulation intervention and (2) Use of PBPK modeling approach to predict and mitigate such interaction. Three drugs were selected namely dasatinib, bosutinib, and gefitinib for which label administration clearly indicates avoidance or staggering with ARA agents. While dasatinib and gefitinib represent the worst case in the case of ARA DDI (>35% reduction in AUC), bosutinib represents milder interaction (17% reduction in AUC). Citric acid has been chosen as an acidifying agent to provide microenvironment and subsequent enhancement in solubility. Although there are no formulations made in the current study, the impact of citric acid on the solubilization behavior was performed through solubility experiments. Considering the average tablet weight of 500 mg with citric acid amount around 10% w/w in the formulation, the concentration of citric acid in the gastric fluid is calculated to be around 0.2 mg/ml considering 250 ml volume. As the pH-dependent DDIs mainly happens at stomach condition, this concentration is deemed to be relevant from in vivo perspective. This concentration was used in the solubility experiments to evaluate the impact of citric acid on the solubility enhancement of the studied drugs. As indicated in Table 3, citric acid significantly enhanced the solubility especially in the range of pH 4.5–6.8 thereby confirming hypothesis of acidic microenvironment. To further evaluate the impact of the enhanced solubility on the in vivo behavior, PBBM were developed for all the studied drugs (Fig. 1). Using the approach mentioned in Figure 1, all the models were successfully developed in the present work.
Physiological models were successfully developed for all the cases using the physicochemical properties (Tables 2 and 3). Elimination parameters were defined appropriately for bosutinib and gefitinib using intravenous data whereas oral data after bioavailability correction was used in case of dasatinib. Similar approach was followed for dasatinib by Heimbach et al. [63] wherein elimination parameters were determined for dasatinib after correcting the bioavailability. The developed models were successfully validated against both intravenous as well as oral pharmacokinetic data and the PEs and folds error were within acceptable limits (Figs. 2 and 3; Table 4). As per the data presented in Figures 2 and 3, the model validation was deemed to be successful. Further, the model’s capability to predict the clinically reported ARA DDI were evaluated and by altering the stomach pH to 5 in the physiology, the models predicted the DDI that are closer to the observed clinical data as per label (Table 1) for all the three drugs. Further, using the enhanced solubility in presence of citric acid, the models were further evaluated to mitigate the ARA impact for all the selected drugs. It is evident from the simulations that due to enhancement in solubility in the range of pH 4.5–6.8, the models for all the studied drugs demonstrated almost nullified ARA DDI (Fig. 5). The outcome from Figure 5 indicates that there is a good probability of nullification of DDI arising from TKI and DDI with introduction of acidifying agent in the formulation. From the in vivo dissolution profiles (Fig. 6) and from the PSA of precipitation time (Fig. 7), it is evident that enhanced solubility in the presence of citric acid has reduced the precipitation and nullified the in vivo dissolution difference between normal stomach (pH 1.3) and altered physiology representing ARA administration (pH 5). The data in Figures 6 and 7 indicates that there is possibility of reduced precipitation and enhanced in vivo dissolution with help of citric acid. Thus, using the acidifying microenvironment, the absence of ARA impact is demonstrated for all the studied drugs. Overall, this work demonstrates the use of formulation intervention and PBBM to mitigate the DDI between ARA and TKI.
 | Figure 7. Parametric sensitivity analysis (precipitation time vs. AUC) (a) and (c) dasatinib oral 100 mg with and without citric acid (b) and (d) bosutinib oral 400 mg with and without citric acid (c) and (e) gefitinib oral 250 mg with and without citric acid. Horizontal access: precipitation time, vertical access: predicted parameter value. [Click here to view] |
We feel that our work is very significant in dealing with the pertinent problem of DDI between ARA and TKIs. As indicated in previous sections, avoidance of ARA is completely not possible during the treatment to avoid gastric events as well as to prevent mucosal damage. Thus, the only possibility is to add acidifying agents in the formulation to mitigate the ARA impact. Although our work achieved its required objective, there are few limitations of the present work that the simulations are based on the in vitro solubility data but not based on the actual formulation that is manufactured with citric acid. To further extend this work, we plan to manufacture actual formulations with citric acid in the future and evaluate them in human study to support the observations in this work and the simulations performed. Further, there are few dissolution medias identified that mimic ARA administration in the fasting condition [64–66]. We further plan to evaluate the prepared formulations in these medias and subsequently shall input in the developed PBBM together with solubility to strengthen the prediction outcomes. Further, model-based predictions are always associated with in vivo variability. In the current modeling exercise, we could not perform population simulations with variability due to unavailability of individual subject data. Once the human study results become available, the variability (e.g., gastric pH variations) can be incorporated into the model to obtain variability in the predictions. Further, development of formulations with acidifying agents is possible and one of such reported formulation is palbociclib tablets wherein succinic acid is included in tablet to provide acidic microenvironment, thereby circumventing impact of ARAs. In the present case as well, for the selected agents, manufacturing formulations with citric acid is feasible and the stability can be evaluated subsequently [67]. We believe that the above-mentioned subsequent research in this area will complement observations in our article and we certainly plan to publish our additional research findings in future publications.
CONCLUSION
The work presented in this article demonstrates the utility of PBBM in predicting and circumventing the DDI arising from TKI and ARA interactions. Three TKIs namely dasatinib, bosutinib, and gefitinib were selected in this study, whose labels indicated restrictions with respect to ARA administration due to clinically relevant DDIs. The PBBM for all the drugs was developed and validated successfully across the formulations and doses. Further, the predictability of PBBM to simulate the observed clinical DDI was demonstrated through appropriate physiological modifications in the stomach. Further, the addition of citric acid to successfully circumvent the ARA TKI DDI was demonstrated using the developed models. Citric acid provided microenvironmental pH and subsequently reduced the precipitation and thus nullified in vivo dissolution differences between pH 1.3 and 5 thereby indicating the possibility of nullifying DDI. Overall, this work demonstrated approaches to mitigate the pertinent challenge of TKI administration, i.e., DDI with ARA and provided a way forward for successful formulation intervention. Such model-based approaches aid significant value to drug development by speeding up the formulation development and avoidance of unnecessary human clinical studies. Further areas of research include in vitro dissolution evaluation to confirm the role of citric acid and further final confirmatory human clinical study to relax the label recommendations with respect to ARA administration.
ACKNOWLEDGMENTS
The authors would like to thank Mahendra Chougule and Ketki Gatade for their contributions to this work.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to the conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agreed to be accountable for all aspects of the work. All the authors are eligible to be authors as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
FINANCIAL SUPPORT
There is no funding to report.
CONFLICT OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVAL
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included in this research article.
PUBLISHER’S NOTE
All claims expressed in this article are solely those of the authors and do not necessarily represent those of the publisher, the editors and the reviewers. This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
USE OF ARTIFICIAL INTELLIGENCE (AI)-ASSISTED TECHNOLOGY
The authors declares that they have not used artificial intelligence (AI)-tools for writing and editing of the manuscript, and no images were manipulated using AI.
REFERENCES
1. Li J, Kuang X. Global cancer statistics of young adults and its changes in the past decade: incidence and mortality from GLOBOCAN 2022. Public Health. 2024;237:336–43.
2. Fidler MM, Gupta S, Soerjomataram I, Ferlay J, Steliarova-Foucher E, Bray F. Cancer incidence and mortality among young adults aged 20–39 years worldwide in 2012: a population-based study. Lancet Oncol. 2017;18:1579–89.
3. Bray F, Laversanne M, Sung H, Ferlay J, Siegel RL, Soerjomataram I, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74:229–63. doi: https://doi.org/10.3322/caac.21834
4. Weeden CE, Hill W, Lim EL, Grönroos E, Swanton C. Impact of risk factors on early cancer evolution. Cell. 2023;186:1541–63. Available from: http://www.cell.com/article/S009286742300274X/fulltext
5. Doll R, Peto R. The causes of cancer: quantitative estimates of avoidable risks of cancer in the United States today. J Natl Cancer Inst. 1981;66:1192–308. doi: CrossRef
6. Li Q, Geng S, Luo H, Wang W, Mo Y-Q, Luo Q, et al. Signaling pathways involved in colorectal cancer: pathogenesis and targeted therapy. Signal Transduct Target Ther. 2024;9(1):1–48. Available from: https://www.nature.com/articles/s41392-024-01953-7
7. Kinnel B, Singh SK, Oprea-Ilies G, Singh R. Targeted therapy and mechanisms of drug resistance in breast cancer. Cancers. 2023;15:1320. Available from: https://www.mdpi.com/2072-6694/15/4/1320/htm
8. Li S, de Camargo Correia GS, Wang J, Manochakian R, Zhao Y, Lou Y. Emerging targeted therapies in advanced non-small-cell lung cancer. Cancers. 2023;15:2899. Available from: https://www.mdpi.com/2072-6694/15/11/2899/htm
9. Huang L, Jiang S, Shi Y. Tyrosine kinase inhibitors for solid tumors in the past 20 years (2001–2020). J Hematol Oncol. 2020;13:1–23. doi: CrossRef
10. Terada T, Noda S, Inui KI. Management of dose variability and side effects for individualized cancer pharmacotherapy with tyrosine kinase inhibitors. Pharmacol Ther. 2015;152:125–34.
11. Yang Y, Li S, Wang Y, Zhao Y, Li Q. Protein tyrosine kinase inhibitor resistance in malignant tumors: molecular mechanisms and future perspective. Signal Transduct Target Ther. 2022;7:1–36. Available from: https://www.nature.com/articles/s41392-022-01168-8
12. Schlam I, Swain SM. HER2-positive breast cancer and tyrosine kinase inhibitors: the time is now. NPJ Breast Cancer 2021;7:1–12. Available from: https://www.nature.com/articles/s41523-021-00265-1
13. Kollipara S, Chougule M, Boddu R, Bhatia A, Ahmed T. Playing hide-and-seek with tyrosine kinase inhibitors: can we overcome administration challenges? AAPS J. 2024;26:1–19. doi: CrossRef
14. Robinson DR, Wu YM, Lin SF. The protein tyrosine kinase family of the human genome. Oncogene. 2000;19:5548–57. Available from: https://www.nature.com/articles/1203957
15. Selick HE, Beresford AP, Tarbit MH. The emerging importance of predictive ADME simulation in drug discovery. Drug Discov Today. 2002;7:109–16.
16. Vinarov Z, Abdallah M, Agundez JAG, Allegaert K, Basit AW, Braeckmans M, et al. Impact of gastrointestinal tract variability on oral drug absorption and pharmacokinetics: an UNGAP review. Eur J Pharm Sci. 2021;162:105812.
17. Cheng F, Li Q, Cui Z, Hong M, Li W, Zhang Y. Dose optimization strategy of the tyrosine kinase inhibitors imatinib, dasatinib, and nilotinib for chronic myeloid leukemia: from clinical trials to real-life settings. Front Oncol. 2023;13:1146108.
18. Gerritse SL, Janssen JBE, Labots M, de Vries R, Rudek M, Carducci M, et al. High-dose administration of tyrosine kinase inhibitors to improve clinical benefit: a systematic review. Cancer Treat Rev. 2021;97:102171.
19. Kang SP, Ratain MJ. Inconsistent labeling of food effect for oral agents across therapeutic areas: differences between oncology and non-oncology products. Clin Cancer Res. 2010;16:4446–51. Available from: /clincancerres/article/16/17/4446/11678/Inconsistent-Labeling-of-Food-Effect-for-Oral
20. Haddad FG, Nasnas C, Sasaki K, Paul S, Issa GC, Rausch C, et al. Using proton pump inhibitors is not associated with adverse outcomes in patients with chronic myeloid leukemia treated with dasatinib. Clin Lymphoma Myeloma Leuk. 2024;24:484–7. Available from: http://www.clinical-lymphoma-myeloma-leukemia.com/article/S2152265024000922/fulltext
21. van Leeuwen RWF, Jansman FGA, Hunfeld NG, Peric R, Reyners AKL, Imholz ALT, et al. Tyrosine kinase inhibitors and proton pump inhibitors: an evaluation of treatment options. Clin Pharmacokinet. 2017;56:683–8. doi: CrossRef
22. Numico G, Fusco V, Franco P, Roila F. Proton pump inhibitors in cancer patients: how useful they are? A review of the most common indications for their use. Crit Rev Oncol Hematol. 2017;111:144–51.
23. Sharma M, Holmes HM, Mehta HB, Chen H, Aparasu RR, Shih YCT, et al. The concomitant use of tyrosine kinase inhibitors and proton pump inhibitors: prevalence, predictors, and impact on survival and discontinuation of therapy in older adults with cancer. Cancer. 2019;125:1155–62. doi: CrossRef
24. Hofmann J, Bart?n?k A, Hauser T, Sedmak G, Beránek J, Ryšánek P, et al. Dasatinib anhydrate containing oral formulation improves variability and bioavailability in humans. Leukemia. 2023;37:2486–92. Available from: https://www.nature.com/articles/s41375-023-02045-1
25. Kollipara S, Ahmed T, Praveen S. Physiologically based pharmacokinetic modelling to predict drug–drug interactions for encorafenib. Part I. Model building, validation, and prospective predictions with enzyme inhibitors, inducers, and transporter inhibitors. Xenobiotica. 2023;53:366–81. doi: CrossRef
26. Kollipara S, Ahmed T, Praveen S. Physiologically based pharmacokinetic modeling (PBPK) to predict drug-drug interactions for encorafenib. Part II. Prospective predictions in hepatic and renal impaired populations with clinical inhibitors and inducers. Xenobiotica. 2023;53:339–56. doi: CrossRef
27. Saeheng T, Na-Bangchang K, Karbwang J. Utility of physiologically based pharmacokinetic (PBPK) modeling in oncology drug development and its accuracy: a systematic review. Eur J Clin Pharmacol. 2018;74:1365–76. doi: CrossRef
28. Boddu R, Kollipara S, Kambam V, Khan SM, Behera S, Murty NNVVSSN, et al. Novel omeprazole delayed release orally disintegrating tablets for enhanced patient compliance: a case of model informed formulation development. Xenobiotica. 2024;54(9):629–41. doi: CrossRef
29. Kollipara S, Martins FS, Sanghavi M, Santos GML, Saini A, Ahmed T. Role of physiologically based biopharmaceutics modeling (PBBM) in fed bioequivalence study waivers: regulatory outlook, case studies and future perspectives. J Pharm Sci. 2024;113:345–58.
30. Bhattiprolu AK, Kollipara S, Boddu R, Arumugam A, Khan SM, Ahmed T. A semi-mechanistic physiologically based biopharmaceutics model to describe complex and saturable absorption of metformin: justification of dissolution specifications for extended release formulation. AAPS PharmSciTech. 2024;25:1–19. doi: CrossRef
31. Kollipara S, Prabhat PK, Saha P, Gupta S, Naidu VR, Ahmed T. Physiologically based biopharmaceutics modeling coupled with biopredictive dissolution in development of bioequivalent formulation for mesalamine enteric coated tablet: a tough nut to crack. AAPS PharmSciTech. 2025;26:1–19. doi: CrossRef
32. Reagents: buffer solutions [Internet]. [cited 2024 Dec 21]. Available from: http://www.uspbpep.com/usp29/v29240/usp29nf24s0_ris1s119.html
33. Kovar C, Loer HLH, Rüdesheim S, Fuhr LM, Marok FZ, Selzer D, et al. A physiologically-based pharmacokinetic precision dosing approach to manage dasatinib drug-drug interactions. CPT Pharmacometrics Syst Pharmacol. 2024;13:1144–59. Available from: https://pubmed.ncbi.nlm.nih.gov/38693610/
34. Abbas R, Leister C, Sonnichsen D. A clinical study to examine the potential effect of lansoprazole on the pharmacokinetics of bosutinib when administered concomitantly to healthy subjects. Clin Drug Investig. 2013;33:589–95. doi: CrossRef
35. Yamazaki S, Loi CM, Kimoto E, Costales C, Varma M V. Application of physiologically based pharmacokinetic modeling in understanding bosutinib drug-drug interactions: importance of intestinal P-glycoprotein. Drug Metab Dispos. 2018;46:1200–11. Available from: https://pubmed.ncbi.nlm.nih.gov/29739809/
36. Abbas R, Chalon S, Leister C, Gaaloul M El, Sonnichsen D. Evaluation of the pharmacokinetics and safety of bosutinib in patients with chronic hepatic impairment and matched healthy subjects. Cancer Chemother Pharmacol. 2013;71:123–32. doi: CrossRef
37. Hsyu PH, Pignataro DS, Matschke K. Absolute bioavailability of bosutinib in healthy subjects from an open-label, randomized, 2-period crossover study. Clin Pharmacol Drug Dev. 2018;7:373–81. doi: CrossRef
38. Swaisland HC, Smith RP, Laight A, Kerr DJ, Ranson M, Wilder-Smith CH, et al. Single-dose clinical pharmacokinetic studies of gefitinib. Clin Pharmacokinet. 2005;44:1165–77. doi: CrossRef
39. Heimbach T, Musuamba Tshinanu F, Raines K, Borges L, Kijima S, Malamatari M, et al. PBBM considerations for base models, model validation, and application steps: workshop summary report. Mol Pharm. 2024;21:5353–72. doi: CrossRef
40. Boddu R, Kollipara S, Vijaywargi G, Ahmed T. Power of integrating PBPK with PBBM (PBPK-BM): a single model predicting food effect, gender impact, drug-drug interactions and bioequivalence in fasting & fed conditions. Xenobiotica. 2023;53:260–78. doi: CrossRef
41. Korashy HM, Rahman AFMM, Kassem MG. Dasatinib. Profiles Drug Subst Excip Relat Methodol. 2014;39:205–37.
42. Wu D, Sanghavi M, Kollipara S, Ahmed T, Saini AK, Heimbach T. Physiologically based pharmacokinetics modeling in biopharmaceutics: case studies for establishing the bioequivalence safe space for innovator and generic drugs. Pharm Res. 2023;40:337–57. doi: CrossRef
43. Xia B, Heimbach T, He H, Lin TH. Nilotinib preclinical pharmacokinetics and practical application toward clinical projections of oral absorption and systemic availability. Biopharm Drug Dispos. 2012;33:536–49. doi: CrossRef
44. FDA. Highlights of prescribing information. [cited 2024 Dec 22]; Available from: www.fda.gov/medwatch
45. FDA, cder. label. [cited 2024 Dec 22]; Available from: www.fda.gov/medwatch
46. FDA, cder. Highlights of prescribing information. [cited 2024 Dec 22]; Available from: www.fda.gov/medwatch
47. Dodd S, Kollipara S, Sanchez-Felix M, Kim H, Meng Q, Beato S, et al. Prediction of ARA/PPI drug-drug interactions at the drug discovery and development interface. J Pharm Sci. 2019;108:87–101.
48. Lewis LD, Koch KM, Reddy NJ, Cohen RB, Lewis NL, Whitehead B, et al. Effects of food on the relative bioavailability of lapatinib in cancer patients. J Clin Oncol. 2009;27:1191–6. doi: CrossRef
49. Xu F, Lee K, Xia W, Liao H, Lu Q, Zhang J, et al. Administration of lapatinib with food increases its plasma concentration in chinese patients with metastatic breast cancer: a prospective phase II study. Oncologist. 2020;25:e1286–91. doi: CrossRef
50. Ferrer F, Tetu P, Dousset L, Lebbe C, Ciccolini J, Combarel D, et al. Tyrosine kinase inhibitors in cancers: treatment optimization – Part II. Crit Rev Oncol Hematol. 2024;200:104385.
51. FDA. Highlights of prescribing information full prescribing information: contents* 1 Indications and usage 2 dosage and administration 2.1 Antidiarrheal prophylaxis 2.2 Recommended dose and schedule 2.3 Dose modifications 3 Dosage forms and strengths 4 Contraindications 5 Warnings and precautions 5.1 Diarrhea 5.2 Hepatotoxicity 5.3 Embryo-fetal toxicity 6 Adverse reactions 6.1 Clinical trials experience 7 Drug interactions 7.1 Effect of other drugs on NERLYNX 7.2 Effect of NERLYNX on other. 2017 [cited 2024 Dec 22]; Available from: www.fda.gov/medwatch
52. FDA. ZELBORAF (vemurafenib) tablet for oral use. [cited 2024 Dec 22]; Available from: www.fda.gov/medwatch
53. Mainie I, Tutuian R, Castell DO. Addition of a H2 receptor antagonist to PPI improves acid control and decreases nocturnal acid breakthrough. J Clin Gastroenterol. 2008;42:676–9. Available from: https://journals.lww.com/jcge/fulltext/2008/07000/addition_of_a_h2_receptor_antagonist_to_ppi.5.aspx
54. Hashimoto H, Kushikata T, Kudo M, Hirota K. Does long-term medication with a proton pump inhibitor induce a tolerance to H2 receptor antagonist? J Gastroenterol. 2007;42:275–8. doi: CrossRef
55. Raoul JL, Guérin-Charbonnel C, Edeline J, Simmet V, Gilabert M, Frenel JS. Prevalence of proton pump inhibitor use among patients with cancer. JAMA Netw Open. 2021;4:e2113739. Available from: https://jamanetwork.com/journals/jamanetworkopen/fullarticle/2781174
56. Uchiyama AAT, Silva PAIA, Lopes MSM, Yen CT, Ricardo ED, Mutão T, et al. Proton pump inhibitors and oncologic treatment efficacy: a practical review of the literature for oncologists. Curr Oncol. 2021;28:783–99. Available from: https://www.mdpi.com/1718-7729/28/1/76/htm
57. Lee JE, Kwon SH, Kwon S, Jung HI, Nam JH, Lee EK. Concomitant use of proton pump inhibitors and palbociclib among patients with breast cancer. JAMA Netw Open. 2023;6:e2324852. Available from: https://jamanetwork.com/journals/jamanetworkopen/fullarticle/2807485
58. Buti S, Tommasi C, Scartabellati G, De Giorgi U, Brighi N, Rebuzzi SE, et al. The impact of proton-pump inhibitors administered with tyrosine kinase inhibitors in patients with metastatic renal cell carcinoma. Anticancer Drugs. 2022;34:178. Available from: https://pmc.ncbi.nlm.nih.gov/articles/PMC9760470/
59. Budha NR, Frymoyer A, Smelick GS, Jin JY, Yago MR, Dresser MJ, et al. Drug absorption interactions between oral targeted anticancer agents and PPIs: is pH-dependent solubility the achilles heel of targeted therapy? Clin Pharmacol Ther. 2012;92:203–13. doi: CrossRef
60. Larfors G, Andersson P, Jesson G, Liljebris C, Brisander M, Lennernäs H, et al. Despite warnings, co-medication with proton pump inhibitors and dasatinib is common in chronic myeloid leukemia, but XS004, a novel oral dasatinib formulation, provides reduced pH-dependence, minimizing undesirable drug–drug interactions. Eur J Haematol. 2023;111:644–54. doi: CrossRef
61. Mitra A, Parrott N, Miller N, Lloyd R, Tistaert C, Heimbach T, et al. Prediction of pH-dependent drug-drug interactions for basic drugs using physiologically based biopharmaceutics modeling: industry case studies. J Pharm Sci. 2020;109:1380–94.
62. Parrott N, Stillhart C, Lindenberg M, Wagner B, Kowalski K, Guerini E, et al. Physiologically based absorption modelling to explore the impact of food and gastric pH changes on the pharmacokinetics of entrectinib. AAPS J. 2020;22:1–13. doi: CrossRef
63. Heimbach T, Kesisoglou F, Novakovic J, Tistaert C, Mueller-Zsigmondy M, Kollipara S, et al. Establishing the bioequivalence safe space for immediate-release oral dosage forms using physiologically based biopharmaceutics modeling (PBBM): case studies. J Pharm Sci. 2021;110:3896–906. Available from: http://jpharmsci.org/article/S002235492100486X/fulltext
64. Segregur D, Flanagan T, Mann J, Moir A, Karlsson EM, Hoch M, et al. Impact of acid-reducing agents on gastrointestinal physiology and design of biorelevant dissolution tests to reflect these changes. J Pharm Sci. 2019;108:3461–77. Available from: http://jpharmsci.org/article/S0022354919304241/fulltext
65. Segregur D, Mann J, Moir A, Karlsson EM, Dressman J. Biorelevant in vitro tools and in silico modeling to assess pH-dependent drug-drug interactions for salts of weak acids: case example potassium raltegravir. J Pharm Sci. 2022;111:517–28. Available from: http://jpharmsci.org/article/S0022354921004858/fulltext
66. Segregur D, Barker R, Mann J, Moir A, Karlsson EM, Turner DB, et al. Evaluating the impact of acid-reducing agents on drug absorption using biorelevant in vitro tools and PBPK modeling - case example dipyridamole. Eur J Pharm Sci. 2021;160:105750.
67. Kuminek G, Salehi N, Waltz NM, Sperry DC, Greenwood DE, Hate SS, et al. Use of gastrointestinal simulator, mass transport analysis, and absorption simulation to investigate the impact of pH modifiers in mitigating weakly basic drugs’ performance issues related to gastric pH: palbociclib case study. Mol Pharm. 2023;20:147–58. doi: CrossRef