INTRODUCTION
Tuberculosis (TB) stands as one of the deadliest diseases in terms of fatalities by infectious disease. Though it is curable and preventable, in 2023, it again became the world’s leading cause of death from a contagious agent after 3 years when it was replaced by COVID-19 [1]. The bacterium responsible for TB, Mycobacterium tuberculosis (MTB), primarily affects the lungs and spreads through the air when an infected person sneezes, coughs, or spits [1–5]. During the latent TB stage within the human body, MTB cells persist in an inactive, nonreplicative state [6], making it challenging to treat. It indicates that if the infection progresses, they are at risk of developing active TB at some point in their lives.
Among the notified TB patients, there are several cases of multidrug-resistant TB and rifampicin-resistant TB. This high number reflects the global challenge of drug resistance, which is more difficult and costly to treat. While there are indications of a slowdown in new TB cases worldwide, the recent rise in drug-resistant rates is alarming and could reverse the positive trends [7]. In response, WHO has adopted a new and comprehensive strategy, the “WHO End TB Strategy,” endorsed by all Member States, guiding concerted efforts at the global, regional, and national levels [8,9].
As resistant forms of TB pose a significant threat to global disease control efforts [10,11], it should be addressed by improving healthcare systems, ensuring proper diagnosis and treatment monitoring, educating patients, and investing in research for new therapies. This underscores a significant progression, starting from the initial discovery of para-amino salicylic acid, followed by isoniazid in the mid-20th century, to the latest discovery of drugs like Bedaquiline and Delamanid [12–15]. Access to drugs such as Bedaquiline and Delamanid remains a challenge in many parts of the world [16]. Therefore, there is a critical need for an improved drug that can simplify the treatment process, reducing treatment duration [17]. A breakthrough for treating drug-susceptible TB includes rifapentine and moxifloxacin, which are of shorter treatment regimens [18,19]. Despite progress, challenges remain, including the need for validated in vitro and animal models that accurately predict the success of new drugs and drug combinations. This gap emphasizes the importance of continued research to enhance our understanding of TB biology and drug efficacy.
The use of computational methodologies has become very important in the discovery and optimization of novel pharmaceutical agents [20–22]. In pharmaceutical research, the utilization of these methods allows researchers to screen large databases of compounds or simulate molecular interactions rapidly. These are also used to optimize drug combinations, particularly important for treating complex diseases like TB or cancer, where multiple drugs may be needed to target different aspects of the disease. By providing detailed insights into drug-target interactions and pharmacokinetic properties before entering clinical trials, computational methodologies help to reduce the risks associated with drug development. Indeed, a first-tier computational screening approach is important to explore the structure–activity relationships among different scaffolds. In-silico studies based on docking performance and quantitative structure-activity relationship (QSAR)-predicted bioactivity at an early stage are needed to identify and prioritize molecular scaffolds for future consideration.
In this study, we collected anti-TB inhibitors from various literature and databases. These compounds were divided into three classes based on their biological activity. The geometries of the compounds were optimized using the density functional theory (DFT) functional, followed by molecular docking calculations to determine docking fitness-based descriptors [23]. DFT-based descriptors, including fragment-based, quantum chemical, thermodynamic, electrostatic, topological, and geometric descriptors, were calculated for structural and statistical analyses. We performed QSAR analysis to predict activities of anti-TB inhibitors, and these models established a relation between the activity of the compounds and the molecular structure descriptors. It is not a complete drug development pipeline. In this study, it is intended as a first-tier computational screening to focus on molecular structure and activity relationships among the selected compounds. Here, scaffolds are prioritized for further synthesis and biological evaluation.
METHODOLOGY
Geometry optimization
Approximately 792 anti-TB inhibitors of various structural analogues were collected from the literature, along with their experimentally derived biological activities measured by minimum inhibitor concentration (MIC50) and inhibitor concentration (IC50) values [24–49]. The geometries of all the considered inhibitors were optimized by DFT functional using hybrid three-parameter Becke-Lee-Yang-Parr (B3LYP) and hybrid meta exchange-correlation functionals (M06) along with 6-31G (d) basis set using the Gaussian 09 program [50]. Frequency calculations were also carried out at the same level of theory to ensure that the obtained stationary points correspond to a minimum on the potential energy surface. The optimized structures were then utilized for docking calculations and QSAR analysis.
Preparation of the dataset
The original IC50 and MIC50 values were transformed into their corresponding pIC50 and pMIC50 values (for the ease of calculations) by applying the formula pMIC = −log MIC and are reported in Tables S1–S14. These data sets include anti-TB inhibitors that exhibit inhibitory potency against 14 distinct cell lines, including pKi, H37rv, Human, Ribosome, Iron_Deficiency, LORA, MABA, Newman, VERO, 7H12, ICB59, LM13, MABA_GAS, and RV128. All the collected anti-TB inhibitors were organized and categorized into three data sets based on pIC50 and pMIC50 values. Four cell lines were chosen for pIC50, four cell lines were selected for pMIC50, and six cell lines were picked for both pIC50 and pMIC50.
Molecular docking
In this investigation, molecular docking calculations were carried out using the GOLD and AutoDock4 programs [51,52]. Here, we have selected 11 crystal structures [Protein Data Bank (PDB) IDs: 1DF7, 1G3U, 1MDB, 1UZN, 1ZID, 2AQK, 2CIB, 2DFT, 2FUM, 3ORM, and 4U0J] to use as receptors in the docking calculations [53–63]. These receptors are known to be specific targets for binding to anti-TB inhibitors. The docking program was utilized to predict the binding affinity and the preferred orientation of the ligands at the receptor binding site. The AutoDock4 tool was used to prepare the receptor structures by adding polar hydrogens, assigning partial and Gasteiger charges, and removing all water molecules and heteroatoms. The binding site of the receptors was defined using a grid of interacting points. The grid points were determined based on the position of the pre-bound ligand within the binding site of each receptor, as observed in its crystal structure. The default parameters of the free energy scoring function were used to assess the binding affinity of the ligand to the receptors.
QSAR
The Comprehensive Descriptors for Structural and Statistical Analysis (CODESSA 3.0) program [64] was used for the generation of the QSAR models. This program calculates various structural and statistical parameters based on 2D and 3D molecular descriptors of the chemical compounds, including fragment-based, thermodynamics, topological, electrostatics, quantum chemical, constitutional, and geometrical descriptors [65]. The DFT-based descriptors are based on the electron density and eigen values of the frontier orbitals and are convenient to describe the chemical reactivity. We have also calculated conceptual DFT-based descriptors, such as hardness, softness, electronegativity, chemical potential, electrophilicity index, and so on, for the compounds and were added them for QSAR modeling [66,67]. The docking scores and other parameters, including protein-ligand hydrogen bond energy (external H-bonds), protein-ligand van der Waals energy (external vdw), sum of the internal torsion and internal vdw terms [internal energy term S (int)], and so on, were also incorporated externally in the database of QSAR modeling to include the information from ligand-receptor interaction. All possible combinations of various descriptors and docking parameters were tested to select the best descriptor-based models. The best multiparameter regression was achieved by iteratively adding descriptors to obtain the best values of statistical criteria such as the highest values of R2, cross-validated R2cv, and the F value. The validation of the generated QSAR models was achieved by dividing the data sets into training and test sets.
RESULTS AND DISCUSSION
Docking analysis
Molecular docking results provide valuable insights into ligand binding at receptor sites through nonbonding interactions with optimal docking scores. The selected inhibitors were subjected to molecular docking calculations using 11 anti-TB receptors (PDB IDs: 1DF7, 1G3U, 1MDB, 1UZN, 1ZID, 2AQK, 2CIB, 2DFT, 2FUM, 3ORM, and 4U0J). Thirty docked poses for each of the inhibitors were generated using each of the receptors. All 792 inhibitors were successfully docked into the 11 receptors. Hydrogen bond interaction, van der Waals interaction, and other weak noncovalent interactions are found to play a major role in binding the inhibitors with the receptors. Many of the inhibitors exhibited good docking scores; however, the docking calculations using 1UZN and 2FUM were unsuccessful for several compounds. The highest-scoring docked poses from receptors 2CIB, 1ZID, and 4U0J showed the best comparability between experimental and predicted activity values. As a result, receptors 2CIB and 1ZID were selected for further analysis. The evaluation of the top-ranked docked poses was based on the analysis of ligand interaction diagrams. Table 1 presents ligand interaction diagrams for selected inhibitors, summarizing their interactions with the receptors based on the highest-scored docked poses. Notably, GLY14 residue displayed the highest number of interactions, followed by GLY96, ALA98, ILE122, and MET147, indicating their primary involvement in interactions with the 2CIB and 1ZID receptors. Receptors 2DFT, 2FUM, and 1G3U were generally associated with hydrogen bonding, while stacking interactions involved ARG153 and LYS13. Additionally, various types of ligand interactions were found to be crucial for binding, with residues ALA22, ASP148, ASP94, MET61, and ILE201 typically involved in these interactions.
Table 1. Ligand interaction diagram for selected receptors.
| PDB ID | Docked pose | Ligand interaction diagram |
|---|---|---|
| 1DF7 | 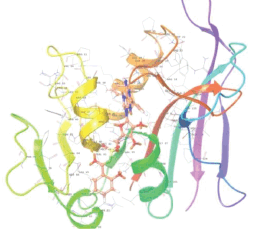 | 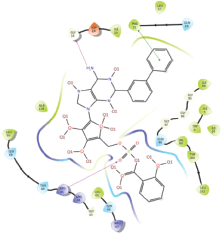 |
| 1G3U | 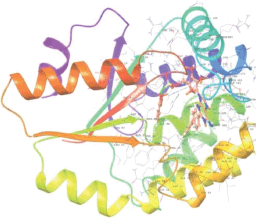 | 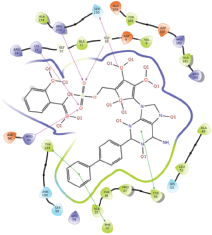 |
| 1MDB | 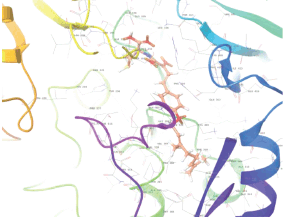 | 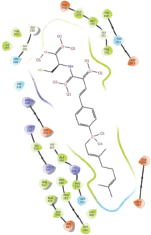 |
| 1UZN | 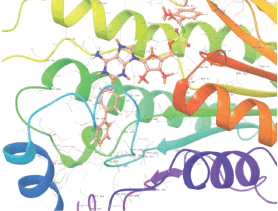 | 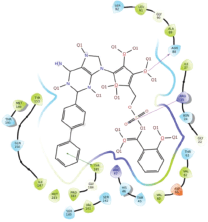 |
| 1ZID | 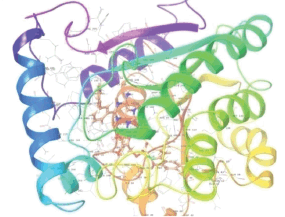 | 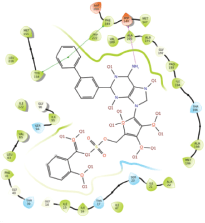 |
| 2AQK | 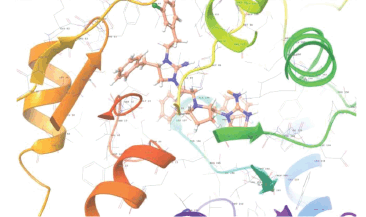 | 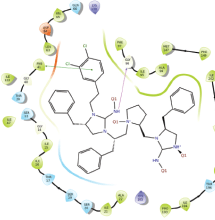 |
| 2CIB | 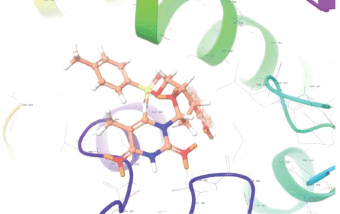 | 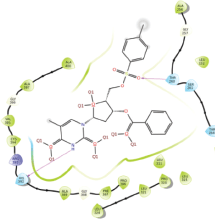 |
| 2DFT | 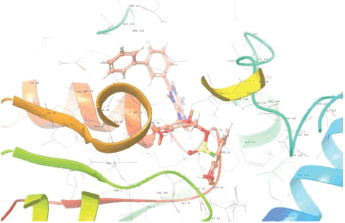 | 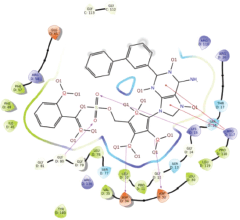 |
| 2FUM | 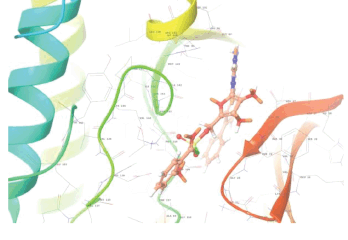 | 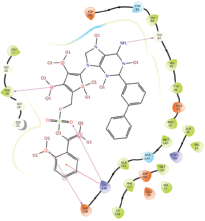 |
| 4FBT | 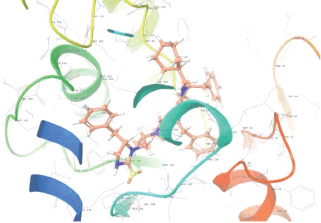 | 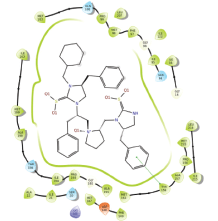 |
| 4U0J | 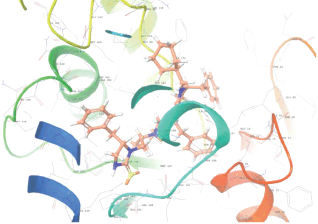 | 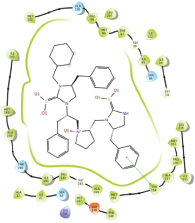 |
QSAR analysis
The CODESSA program was used to generate QSAR models (Set 1–14) using both conventional and conceptual descriptors, along with the docking scores of all considered anti-TB inhibitors. Multiple linear regressions (MLRs) analysis was performed on all possible combinations of descriptors. Traditional descriptors, such as constitutional, geometrical, topological, electrostatic, and quantum chemical features, were also computed. Conceptual DFT-based descriptors, including hardness, chemical potential, and electrophilicity index, were incorporated externally. In addition, docking-based descriptors, including docking fitness score, H-bond, and van der Waals interactions, obtained from docking results, were also incorporated into QSAR modeling. We have used various numbers of descriptors in regression analyses to ensure the mathematical significance and robustness of the calculations. Table S15 represents the selected descriptors used to generate the QSAR models.
The optimal number of descriptors for the final QSAR model has been determined. A summary of the investigation for various descriptor combinations is presented in Table 2, along with the important measurable parameters associated with each model. All the generated models were ranked according to their R2 (correlation coefficient), R2cv (cross-validated R2), F (Fischer statistic), s2 (SD), and SE values. The inhibitors were divided into a training and a test set for the rigorous validation of the generated QSAR models. The descriptors and predicted activities derived from MLR and cross-validation of the regression coefficient (cv) are presented in Table 2 for both the training and test sets.
Table 2. Statistical significances of the selected QSAR models for different sets of compounds.
| Set | #Comp. | #Des. | R2 | R2cv | SE | S2 | F | Regression equation for descriptor-based models | CDFTdescriptors |
|---|---|---|---|---|---|---|---|---|---|
| Set 1 pIC50_pKi | 47 (42) 5* | 7 | 0.70 (0.71) | 0.52 (0.54) | 0.38 (0.37) | 0.18 (0.17) | 10.43 (11.66) | =D24 (−0.11) + D76 (−2.05) + D41 (151.16) + D8 (24.43) + D72 (−11.33) + D68 (9.37) + D19 (−0.42) + (−176.19) | Y |
| Set 2 pIC50_H37rv | 111 (102) 9* | 10 | 0.63 (0.64) | 0.55 (0.56) | 0.68 (0.68) | 0.52 (0.52) | 16.68 (15.80) | =D39 (−80.96) + D25 (32.01) + D20 (0.92) + D5 (−57.37) + D40 (4.54) + D74 (0.23) + D45 (0.06) + D57 (1.58) + D7 (−8.84) + D52 (−0.71) + 166.06 | Y |
| Set 3 pIC50_Human | 53 (46) 7* | 6 | 0.81 (0.81) | 0.76 (0.76) | 0.53 (0.52) | 0.33 (0.32) | 33.63 (28.32) | =D63 (−0.28) + D22 (−39.37) + D37 (2.88) + D41 (59.25) + D28 (−0.16) + D61 (0.24) + (−56.10) | Y |
| Set 4 pIC50_Ribosome | 52 (47) 5* | 6 | 0.70 (0.73) | 0.56 (0.54) | 0.30 (0.26) | 0.10 (0.08) | 17.16 (17.72) | =D29 (−0.38) + D30 (0.06) + D67 (0.43) + D77 (10.76) + D60 (0.00) + D13 (3.72) + (−2.33) | Y |
| Set 5 pMIC50_Iron_pKi | 24 (20) 4* | 5 | 0.88 (0.88) | 0.78 (0.79) | 0.43 (0.41) | 0.25 (0.25) | 25.57 (20.70) | =D73 (−36.76) + D71 (0.00) + D21 (498.75) + D4 (−133.36) + D33 (0.65) + 236.00 | Y |
| Set 6 pMIC50_LORA | 110 (100) 10* | 8 | 0.77 (0.68) | 0.73 (0.62) | 0.29 (0.35) | 0.09 (0.13) | 41.80 (24.63) | =D9 (2.23) + D46 (−0.09) + D50(−0.09) + D49 (0.09) + D18 (0.32) + D12 (11.58) + D13 (0.55) + D73 (−3.30) + 0.80 | Y |
| Set 7 pMIC50_MABA | 130 (120) 10* | 10 | 0.81 (0.80) | 0.77 (0.75) | 0.65 (0.67) | 0.47 (0.49) | 49.15 (52.51) | =D11 (46.31) + D38 (−60.35) + D23 (−0.38) + D65 (−0.25) + D53 (0.46) + D14 (−0.01) + D36 (29.64) + D42 (−9.82) + D3 (15.08) + D35 (27.42) + (−77.42) | N |
| Set 8 pMIC50_Newman | 28 (24) 4* | 5 | 0.94 (0.94) | 0.91 (0.90) | 0.14 (0.14) | 0.02 (0.03) | 74.43 (58.18) | =D62 (0.24) + D64 (−0.69) + D61 (0.61) + D32 (0.30) + D2 (0.33) + (−0.23) | Y |
| Set 9 pIC50_VERO_Cell | 74 (71) 3* | 7 | 0.98 (0.98) | 0.98 (0.98) | 0.10 (0.10) | 0.01 (0.01) | 699 (637) | =D6 (19.16) + D55 (1.95) + D61 (0.07) + D27 (−0.05) + D44 (0.01) + D20 (0.00) + D51 (0.04) + (−22.34) | Y |
| Set 10 pMIC50_7H12 | 17 (14) 3* | 2 | 0.64 (0.78) | 0.56 (0.64) | 0.46 (0.38) | 0.26 (0.18) | 12.23 (19.96) | =D1 (0.52) + D31 (0.12) + 4.95 | Y |
| Set 11 pMIC50_ICB59 | 42 (37) 5* | 5 | 0.61 (0.63) | 0.49 (0.51) | 0.40 (0.37) | 0.18 (0.16) | 11.12 (11.18) | =D69 (−0.03) + D32 (−1.19) + D56 (0.30) + D16 (0.99) + D70 (0.07) + 1.41 | Y |
| Set 12 pMIC50_LM13 | 36 (30) 6* | 5 | 0.65 (0.66) | 0.51 (0.51) | 0.32 (0.30) | 0.12 (0.12) | 9.05 (7.53) | =D21 (−182.71) + D29 (0.39) + D48 (0.06) + D26 (0.12) + D15 (0.03) + D66 (−0.38) + (−9.46) | Y |
| Set 13 pMIC50_MABA_GAS | 16 (13) 3* | 2 | 0.88 (0.86) | 0.80 (0.77) | 0.37 (0.34) | 0.17 (0.15) | 48.05 (31.91) | =D75 (−61.89) + D27 (−0.24) + 4.90 | Y |
| Set 14 pMIC50_RV128 | 52 (47) 5* | 6 | 0.76 (0.76) | 0.69 (0.68) | 0.29 (0.28) | 0.09 (0.09) | 24.72 (21.81) | =D70 (−0.09) + D17 (−1.46) + D36 (18.13) + D10 (3.28) + D23 (−2.10) + D43 (1.59) + (−8.19) | Y |
#Comp is number of compounds; #Des is number of descriptors; R2 is correlation coefficient; R2cv is cross-validation coefficient; s2 is SD; F is Fischer statistics; numbers in italics is the number of considered compounds; numbers in brackets is after division in training and test sets; * shows the numbers of compounds in the test set.
Three classes of inhibitors
Class 1: for cell lines
In class 1, Sets 1, 2, 3, and 4 comprise a total of 47, 111, 53, and 52 compounds, respectively. These sets pertain to TB-infected cells from pki, H37Rv, human, and ribosome. The models for these sets, using 7, 10, 6, and 6 descriptors, respectively, are highlighted to demonstrate the optimal models.
To validate these QSAR models, a division into training and test sets was implemented. Test sets consisted of 5, 9, 7, and 5 randomly selected inhibitors, while the remaining 42, 102, 46, and 47 inhibitors were allocated to training sets. Statistical parameters for both models, with and without test set division, are given in Table 2.
The combination of the selected descriptors exhibited notable R2 (0.70) and R2cv (0.52) for Set 1, R2 (0.63) and R2cv (0.55) for Set 2, R2 (0.81) and R2cv (0.76) for Set 3, and R2 (0.70) and R2cv(0.56) for Set 4. The comparability between experimental and predicted values of pIC50 is depicted in Figure 1, with test set inhibitors highlighted in red. These plots affirm the validity of predicting test set inhibitors, indicating the utility of these models for unknown compounds. Tables S1–S4 represent the detailed experimental and calculated bioactivity values of class 1 set inhibitors. These results show comparability of the predicted activity values obtained from the QSAR models with the experimentally available values.
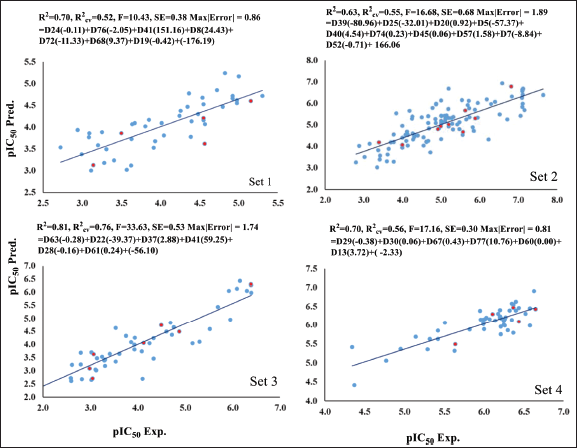 | Figure 1. The plot of experimental (pIC50 Exp.) versus predicted (pIC50 Pred.) bioactivity values for Set 1–4. Blue points show training set compounds, and red points show test set compounds. [Click here to view] |
Class 2: for cell lines
Table 2 presents the results for four different sets of anti-TB-infected cells: Set 5, 6, 7, and 8. Set 5 comprises 24 compounds with five descriptors. This set is divided into a training set of 20 compounds and a test set of four compounds, yielding R2 values of 0.88 and R2cv values of 0.78. Set 6 consists of 110 compounds with eight descriptors, divided into a training set of 100 compounds and a test set of 10 compounds, resulting in R2 and R2cv values of 0.77 and 0.73, respectively. Similarly, Set 7 includes 130 compounds with 10 descriptors. This set is divided into a training set of 120 compounds and a test set of 10 compounds, leading to R2 and R2cv values of 0.81 and 0.77, respectively. Finally, Set 8 comprises 28 compounds with five descriptors. This set is divided into a training set of 24 compounds and a test set of four compounds. The statistical parameters R2 and R2cv values for Set 8 are 0.94 and 0.91, respectively. Integrating the statistical information from all these descriptors enhances the overall quality of the models used for the test set division. Notably, the positive contributions from DFT and docking-based descriptors complement the coefficient values obtained from the conventional QSAR-based descriptor model across all sets. Figure 2 shows the plots illustrating the relationship between experimental and predicted values of pMIC50 for all these class 2 sets. Sets 6 and 7 exhibit particularly strong statistical performance, while Sets 5 and 8 also demonstrate very good parameters. For class 2 inhibitors, calculated values of bioactivity derived from the generated models are reported in Tables S5–S8, along with the experimentally determined values, and these results show good comparability.
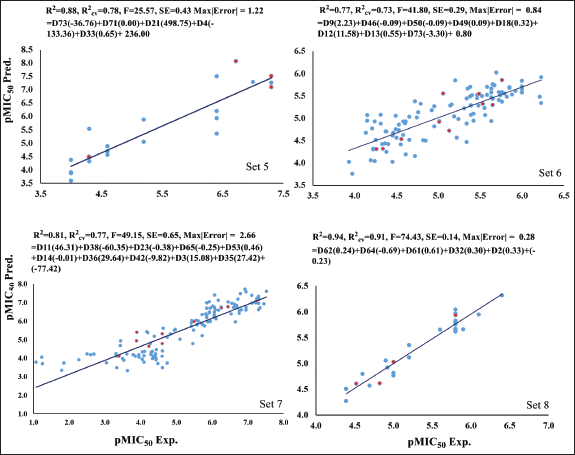 | Figure 2. The plot of experimental (pMIC50 Exp.) versus predicted (pMIC50 Pred.) bioactivity values for Set 5–8. Blue points show training set compounds, and red points show test set compounds. [Click here to view] |
Class 3: for both and cell lines
Sets 9–14, similar to classes 1 and 2, show a strong statistical performance with eight sets of compounds.
Set 9 includes 74 compounds, with 71 in the training set and 3 in the test set, achieving an R2 = 0.98, R2cv = 0.98. Set 10 has 17 compounds, comprising 14 in the training set and 3 in the test set, with an R2 = 0.64, R2cv = 0.56. In Set 11, there are 42 compounds, with 37 in the training set and 5 in the test set, resulting in an R2 = 0.61, R2cv = 0.59. Set 12 contains 36 compounds, 30 of which are in the training set and 6 in the test set, with an R2 = 0.65, R2cv = 0.51. Set 13 consists of 16 compounds, featuring 13 in the training set and 3 in the test set, achieving an R2 = 0.88, R2cv = 0.80. Finally, Set 14 has 52 compounds, with 47 in the training set and 5 in the test set, resulting in an R2 = 0.76, R2cv = 0.69. All of these sets exhibit statistically significant descriptors, as presented in Table 2. For Sets 9–14, the plot between experimental and predicted values of bioactivity is shown in Figure 3.
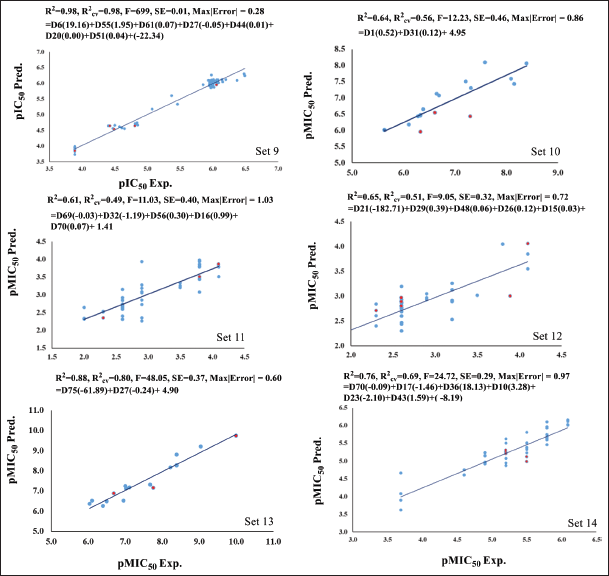 | Figure 3. The plot of experimental (pIC50 Exp. and pMIC50 Exp.) versus predicted (pIC50 Pred. and pMIC50 Pred.) bioactivity values for Set 9–14. Blue points show training set compounds, and red points show test set compounds. [Click here to view] |
Based on these models, we have predicted bioactivity of three classes of compounds, and the detailed predicted values of bioactivity of all the selected anti-TB inhibitors of Set 9–14 are given in Tables S9–S14. The applicability of the generated QSAR models was validated by performing leave-one-out, leave-many-out cross validations, and Y-randomization tests for the prediction of bioactivity of the compounds. The statistical parameters of these models are found to be significant for the class of selected inhibitors.
Moulishankar et al. [68] did the QSAR modeling for a set of 53 novel thiazolidine 4-one derivatives that have the potential to act as potent anti-TB inhibitors. These molecules were collected from the literature with their MIC activity. In their approach, they had predicted anti-TB activity by splitting the molecules into a training set (43 compounds) and a test set (10 compounds). This study represented the value of R2 as 0.9092 and R2adj as 0.8950. In our study, thiazolidine 4-one derivatives with MIC activity were included in Sets 6, 7, 10, 13, and 14, along with other structural entities. These sets exhibit strong statistical performance. Among these, the QSAR model for Set 13 shows a value of R2 as 0.88, which is similar to the model generated by Moulishankar et al. [68] As Set 13 contains thiazolidine 4-one derivatives as well as some more inhibitors; it can be used to predict the bioactivity for a wide range of compounds. Similarly, Valencia et al. [69] designed and tested the bioactivity for a set of novel quinolinone-based thiosemicarbazones. In their QSAR modeling, they achieved the statistical parameters as R2 = 0.83; F = 47.96; s = 0.31. In this present study, a few quinolinone-based thiosemicarbazones have been included in Set 2. The statistical parameters suggest that the model generated by Valencia et al. [69] is specific for quinolinone-based thiosemicarbazones to predict the bioactivity values. Pietra et al. [70] performed the QSAR modeling on purine analogues and reported sensitivity as 0.85 for the training set and 0.82 for the test set. In our study, these types of compounds were included in Set 5 with a good statistical performance of R2 of 0.88. This Set 5 model may be significant for predicting bioactivity for purine derivatives. The QSAR model for Set 8 can also be a good model to predict bioactivity for phenanthrene derivatives with significant statistical parameters. This model shows R2 as 0.94, which is similar to that shown by Shagufta et al. [71] for a series of 44 diaryloxy-methano-phenanthrene derivatives.
CONCLUSION
This investigation employed molecular docking scores and QSAR for the analysis of 792 anti-TB inhibitors. A total of 14 QSAR models were developed using MLR and partial least squares, based on data collected from various biological activities of anti-TB inhibitors. We have generated structure-based QSAR models to predict bioactivity values for a set of anti-TB inhibitors against various cell lines using an appropriate and vital set of descriptors. This was achieved by dividing the data set into training and test sets to fit the models with the validation of leave-one-out and leave-many-out. This study uses conceptual DFT-based descriptors along with conventional descriptors such as constitutional, topological, docking, geometrical, electrostatic, and quantum chemical. The calculated descriptors were based on conceptual DFT, emphasizing hardness, chemical potential, and the electrophilicity index. The predicted bioactivity values show good comparability with the experimentally available values. Some compounds show higher predicted values, which implies the necessity of further research on these compounds. Notably, quantum chemical descriptors emerged as crucial, and the incorporation of conceptual DFT descriptors has significantly improved the statistical parameters of the model across various scenarios.
Limitation
Future studies are needed to include MD simulations to confirm the dynamic stability of top-ranked complexes identified here. Additionally, while this study identifies promising scaffolds based on docking and QSAR models, prioritizing for synthesis; subsequent ADMET profiling is essential before preclinical evaluation.
ACKNOWLEDGMENT
SK, is grateful to the University Grants Commission for financial support through the Dr. D. S. Kothari Postdoctoral Fellowship (No.F.4-2/2006 (BSR)/PH/18-19/0022). SB is thankful to Science & Engineering Research Board, Department of Science and Technology for Financial support through sanction order PDF/2016/002053. We also thanks ‘Param-Ishan Supercomputer’ IIT Guwahati’ and Department of Physics, BHU, Varanasi, for computational facilities.
PREPRINT DISCLOSURE
A portion of this work has been previously made publicly available as a preprint on the ChemRxiv server (DOI: https://doi.org/10.26434/ chemrxiv-2024-czpvq). The preprint version may list a slightly different author lineup due to internal revisions made during manuscript development and submission. The current author list reflects the final contributions and authorship agreed upon for journal submission, in accordance with the guidelines for authorship and contribution. All co-authors have reviewed and approved the submitted version of the manuscript
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included in this research article.
PUBLISHER’S NOTE
All claims expressed in this article are solely those of the authors and do not necessarily represent those of the publisher, the editors and the reviewers. This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
USE OF ARTIFICIAL INTELLIGENCE (AI)-ASSISTED TECHNOLOGY
The authors declares that they have not used artificial intelligence (AI)-tools for writing and editing of the manuscript, and no images were manipulated using AI.
SUPPORTING INFORMATION
Experimental and predicted activity values of studied 792 inhibitors using different descriptor-based QSAR models and a list of selected descriptors utilized for generating the selected QSAR models.
REFERENCES
1. Global tuberculosis report 2024. Geneva, Switzerland: World Health Organization; 2024.
2. Kochi A. The global tuberculosis situation and the new control strategy of the World Health Organization. Tubercle 1991;72(1):1–6. CrossRef
3. Migliori GB, Sotgiu G, Rosales-Klintz SR, van der Werf MJ. European Union standard for tuberculosis care on treatment of multidrug-resistant tuberculosis following new World Health Organization recommendations. Eur Respir J. 2018;52(5):1801617. CrossRef
4. Floyd K, Glaziou P, Zumla A, Raviglione M. The global tuberculosis epidemic and progress in care, prevention, and research: an overview in year 3 of the end TB era. Lancet Respir Med. 2018;6(4):299–314. CrossRef
5. Kochi A. The global tuberculosis situation and the new control strategy of the World Health Organization. Bull World Health Organ. 2001;79(1):71–5.
6. Connolly LE, Edelstein PH, Ramakrishnan L. Why is long-term therapy required to cure tuberculosis? PLoS Med. 2007;4(3):435–42. CrossRef
7. Garcia-Basteiro AL, Brew J, Williams B, Borgdorff M, Cobelens F. What is the true tuberculosis mortality burden? Differences in estimates by the World Health Organization and the Global Burden of Disease study. Int J Epidemiol. 2018;47(5):1549–60. CrossRef
8. Kirigia JM, Muthuri RDK. Productivity losses associated with tuberculosis deaths in the World Health Organization African region. Infect Dis Poverty. 2016;5(1):43. CrossRef
9. Falzon D, Schünemann HJ, Harausz E, González-Angulo L, Lienhardt C, Jaramillo E, et al. World Health Organization treatment guidelines for drug-resistant tuberculosis, 2016 update. Eur Respir J. 2017;49(3):1602308. CrossRef
10. Varaine F, Guglielmetti L, Huerga H, Bonnet M, Kiria N, Sitienei JK, et al. Eligibility for the shorter multidrug-resistant tuberculosis regimen: ambiguities in the World Health Organization recommendations. Am J Respir Crit Care Med. 2016;194(8):1028–9. CrossRef
11. Rado TA, Bates JH, Engel HW, Mankiewicz E, Murohashi T, Mizuguchi Y, et al. World Health Organization studies on bacteriophage-typing of mycobacteria-subdivision of species Mycobacterium tuberculosis. Am Rev Respir Dis. 1975;111(4):459–68.
12. CDC. Extensively drug-resistant tuberculosis—United States, 1993–2006. Morb Mortal Wkly Rep. 2007;56(11):250–3.
13. Young C, Walzl G, Plessis ND. Therapeutic host-directed strategies to improve outcome in tuberculosis. Mucosal Immunol. 2020;13(2):190–204. CrossRef
14. Shahab M, Morais GCF, Akash S, Fulco UL, Oliveira JIN, Zheng G, et al. A robust computational quest: discovering potential hits to improve the treatment of pyrazinamide-resistant Mycobacterium tuberculosis. J Cell Mol Med. 2024;28(8):e18279. CrossRef
15. Halleux CM, Falzon D, Merle C, Jaramillo E, Mirzayev F, Olliaro P, et al. The World Health Organization global aDSM database: generating evidence on the safety of new treatment regimens for drug-resistant tuberculosis. Eur Respir J. 2018;51(3):1701643. CrossRef
16. Capela R, Félix R, Clariano M, Nunes D, Perry MJ, Lopes F. Target identification in anti-tuberculosis drug discovery. Int J Mol Sci. 2023;24(13):10482. CrossRef
17. Perveen S, Kumari D, Singh K, Sharma R. Tuberculosis drug discovery: progression and future interventions in the wake of emerging resistance. Eur J Med Chem. 2022;229:114066. CrossRef
18. Winters N, Butler-Laporte G, Menzies D. Efficacy and safety of World Health Organization group 5 drugs for multidrug-resistant tuberculosis treatment. Eur Respir J. 2015;46(5):1461–70. CrossRef
19. Ciaranello A, Lu Z, Ayaya S, Losina E, Musick B, Vreeman R. Incidence of World Health Organization stage 3 and 4 events, tuberculosis and mortality in untreated, HIV-infected children enrolling in care before 1 year of age AnIeDEA (International Epidemiologic Databases To Evaluate AIDS) East Africa regional analysis. Pediatr Infect Dis J. 2014;33(6):623–9. CrossRef
20. Pingale RN, Pratyush K, Wagh B, Jain AA. Evaluation of PPAR gamma agonists: a molecular docking and QSAR study of chalcone analog. J Appl Pharm Sci. 2025;15(02):155–63. CrossRef
21. Yoosefian M, Moghani MZ, Juan A. In silico evaluation of atazanavir as a potential HIV main protease inhibitor and its comparison with new designed analogs. Comput Biol Med. 2022;145:105523. CrossRef
22. Yoosefian M, Sabaghian H. The role of weak interactions in evaluation of inhibitory potential of Indinavir as an HIV protease inhibitor and its comparison with innovative drug candidates. Comput Biol Med. 2025;187:109675. CrossRef
23. Meng XY, Zhang HX, Mezei M, Cui M. Molecular docking: a powerful approach for structure-based drug discovery. Curr Comput Aided Drug Des. 2011;7(2):146–57. CrossRef
24. Ragno R, Marshall GR, Santo RD, Costi R, Massa S, Rompei R, et al. Antimycobacterial pyrroles: synthesis, anti-Mycobacterium tuberculosis activity and QSAR studies. Bioorg Med Chem. 2000;8(6):1423–32. CrossRef
25. Nayyar A, Monga V, Malde A, Coutinho E, Jain R. Synthesis, anti-tuberculosis activity, and 3D-QSAR study of 4-(adamantan-1-yl)-2-substituted quinolines. Bioorg Med Chem. 2007;15(2):626–40. CrossRef
26. Senthilkumar P, Dinakaran M, Chandraseakaran Y, Yogeeswari P, Sriram D. Synthesis and in-vitro antimycobacterial evaluation of 1-(cyclopropyl/2,4-difluorophenyl/tert-butyl)-1,4-dihydro- 8-methyl-6-nitro-4-oxo-7-(substituted secondary amino)quinoline-3-carboxylic acids. Archiv der Pharmazie (Weinheim) 2009;342(2):100–12. CrossRef
27. Hearn MJ, Cynamon MH, Chen MF, Coppins R, Davis J, Kang HJO, et al. Preparation and antitubercular activities in vitro and in vivo of novel Schiff bases of isoniazid. Eur J Med Chem. 2009;44(10):4169–78. CrossRef
28. Guan YF, Song X, Qiu MH, Luo SH, Wang BJ, Hung NV, et al. Bioassay-guided isolation and structural modification of the anti-TB resorcinols from Ardisia gigantifolia. Chem Biol Drug Des. 2016;88(2):293–301. CrossRef
29. Yoya GK, Bedos-Belval F, Constant P, Duran H, Daffé M, Baltas M. Synthesis and evaluation of a novel series of pseudo-cinnamic derivatives as antituberculosis agents. Bioorg Med Chem Lett. 2009;19(2):341–3. CrossRef
30. De P, Yoya GK, Constant P, Bedos-Belval F, Duran H, SaffonN. Design, synthesis, and biological evaluation of new cinnamic derivatives as antituberculosis agents. J Med Chem. 2011;54(5):1449–61. CrossRef
31. Suresh A, Suresh N, Misra S, Kumar MMK, Venkata K, Sekhar GC. Design, synthesis and biological evaluation of new substituted sulfonamide tetrazole derivatives as antitubercular agents. Chem Select 2016;1(8):1705–10. CrossRef
32. Pitta E, Rogacki MK, Balabon O, Huss S, Cunningham F, Lopez-Roman EM, et al. Searching for new leads for tuberculosis: design, synthesis, and biological evaluation of novel 2-quinolin-4-yloxyacetamides. J Med Chem. 2016;59(14):6709–28. CrossRef
33. Biava M, Porretta GC, Poce G, Deidda D, Pompei R, Tafi A, et al. Antimycobacterial compounds. Optimization of the BM 212 structure, the lead compound for a new pyrrole derivative class. Bioorg Med Chem. 2005;13(4):1221–30. CrossRef
34. Moraski GC, Seeger N, Miller PA, Oliver AG, Boshoff HI, Cho S, et al. Arrival of imidazo[2,1-b]thiazole-5-carboxamides: potent anti-tuberculosis agents that target QcrB. ACS Infect Dis. 2016;2(6):393–8. CrossRef
35. Biava M, Porretta GC, Poce G, De Logu A, Meleddu R, De Rossi E, et al. 1,5-Diaryl-2-ethyl pyrrole derivatives as antimycobacterial agents: design, synthesis, and microbiological evaluation. Eur J Med Chem. 2009;44(11):4734–8. CrossRef
36. Zampieri D, Mamolo MG, Laurini E, Scialino G, Banfi E, Vio L. Antifungal and antimycobacterial activity of 1-(3,5-diaryl-4,5-dihydro-1H-pyrazol-4-yl)-1H-imidazole derivatives. Bioorg Med Chem. 2008;16(8):4516–22. CrossRef
37. Sasaki H, Haraguchi Y, Itotani M, Kuroda H, Hashizume H, Tomishige T, et al. Synthesis and antituberculosis activity of a novel series of optically active 6-nitro-2,3-dihydroimidazo[2,1-b]oxazoles. J Med Chem. 2006;49(26):7854–60. CrossRef
38. Sutherland HS, Blaser A, Kmentova I, Franzblau SG, Wan B, Wang Y, et al. Synthesis and structure-activity relationships of antitubercular 2-nitroimidazooxazines bearing heterocyclic side chains. J Med Chem. 2010;53(2):855–66. CrossRef
39. Thompson AM, O’Connor PD, Blaser A, Yardley V, Maes L, Gupta S, et al. Repositioning antitubercular 6-nitro-2,3-dihydroimidazo[2,1-b][1,3]oxazoles for neglected tropical diseases: structure-activity studies on a preclinical candidate for visceral leishmaniasis. J Med Chem. 2016;59(6):2530–50. CrossRef
40. Koseki Y, Aoki S. Computational medicinal chemistry for rational drug design: identification of novel chemical structures with potential anti-tuberculosis activity. Curr Top Med Chem. 2014;14(1):176–88. CrossRef
41. Kumar M, Singh K, Naran K, Hamzabegovic F, Hoft DF, Warner DF, et al. Design, synthesis, and evaluation of novel hybrid efflux pump inhibitors for use against Mycobacterium tuberculosis. ACS Infect Dis. 2016;2(10):714–25. CrossRef
42. Scotti L, Tullius Scotti M, de Oliveira Lima E, Sobral da Silva M, do Carmo Alves de Lima M, da Rocha Pitta I, et al. Experimental methodologies and evaluations of computer-aided drug design methodologies applied to a series of 2-aminothiophene derivatives with antifungal activities. Molecules 2012;17(3):2298–315. CrossRef
43. Lilienkampf A, Mao J, Wan B, Wang Y, Franzblau SG, Kozikowski AP. Structure-activity relationships for a series of quinoline-based compounds active against replicating and nonreplicating Mycobacterium tuberculosis. J Med Chem. 2009;52(7):2109–18. CrossRef
44. Saquib M, Husain I, Sharma S, Yadav G, Singh VK, Sharma SK, et al. 2,3-dideoxy hex-2-enopyranosid-4-uloses as promising new anti-tubercular agents: design, synthesis, biological evaluation and SAR studies. Eur J Med Chem. 2011;46(6):2217–23. CrossRef
45. Nefzi A, Appel J, Arutyunyan S, Houghten RA. Parallel synthesis of chiral pentaamines and pyrrolidine containing bis-heterocyclic libraries. Multiple scaffolds with multiple building blocks: a double diversity for the identification of new antitubercular compounds. Bioorg Med Chem Lett. 2009;19(17):5169–175. CrossRef
46. Liu ML, Pan XY, Yang T, Zhang WM, Wang TQ, Wang HY, et al. The synthesis and antistaphylococcal activity of dehydroabietic acid derivatives: Modifications at C-12. Bioorg Med Chem Lett. 2016;26(22):5492–6. CrossRef
47. He C, Preiss L, Wang B, Fu L, Wen H, Zhang X, et al. Structural simplification of bedaquiline: the discovery of 3-(4-(N,N-dimethylaminomethyl)phenyl)quinoline-derived antitubercular lead compounds. ChemMedChem. 2017;12(2):106–19. CrossRef
48. Liu J, Bruhn DF, Lee RB, Zheng Z, Janusic T, Scherbakov D, et al. Structure-activity relationships of spectinamide antituberculosis agents: a dissection of ribosomal inhibition and native efflux avoidance contributions. ACS Infect Dis. 2017;3(1):72–88. CrossRef
49. North EJ, Scherman MS, Bruhn DF, Scarborough JS, Maddox MM, Jones V, et al. Design, synthesis and anti-tuberculosis activity of 1-adamantyl-3-heteroaryl ureas with improved in vitro pharmacokinetic properties. Bioorg Med Chem. 2013;21(9):2587–99. CrossRef
50. Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, et al. Gaussian 09, Revision A.02. Wallingford CT: Gaussian, Inc.; 2016.
51. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–61. CrossRef
52. Verdonk ML, Chessari G, Cole JC, Hartshorn MJ, Murray CW, Nissink JW, et al. Modeling water molecules in protein-ligand docking using GOLD. J Med Chem. 2005;48(20):6504–15. CrossRef
53. Li R, Sirawaraporn R, Chitnumsub P, Sirawaraporn W, Wooden J, Athappilly F, et al. Three-dimensional structure of M. tuberculosis dihydrofolate reductase reveals opportunities for the design of novel tuberculosis drugs. J Mol Biol. 2000;295:307–23. CrossRef
54. Li de la Sierra I, Munier-Lehmann H, Gilles AM, Barzu O, Delarue M. X-ray structure of TMP kinase from Mycobacterium tuberculosis complexed with TMP at 1.95 A resolution. J Mol Biol. 2001;311:87–100. CrossRef
55. May JJ, Kessler N, Marahiel MA, Stubbs MT. Crystal structure of DhbE, an archetype for aryl acid activating domains of modular nonribosomal peptide synthetases. Proc Natl Acad Sci USA. 2002;99:12120–5. CrossRef
56. Cohen-Gonsaud M, Ducasse S, Hoh F, Zerbib D, Labesse G, Quemard A. Crystal structure of maba from Mycobacterium tuberculosis, a reductase involved in long-chain fatty acid biosynthesis. J Mol Biol. 2002;320:249. CrossRef
57. Rozwarski DA, Grant GA, Barton DH, Jacobs WR Jr, Sacchettini JC. Modification of the NADH of the isoniazid target (InhA) from Mycobacterium tuberculosis. Science 1998;279:98–102. CrossRef
58. Oliveira JS, Pereira JH, Canduri F, Rodrigues NC, de Souza ON, de Azevedo WF Jr, et al. Crystallographic and pre-steady-state kinetics studies on binding of NADH to wild-type and isoniazid-resistant enoyl-ACP(CoA) reductase enzymes from Mycobacterium tuberculosis. J Mol Biol. 2006;359:646–66. CrossRef
59. Podust LM, Von Kries JP, Eddine AN, Kim Y, Yermalitskaya LV, Kuehne R, et al. Small molecule scaffolds for Cyp51 inhibitors identified by high throughput screening and defined by X-ray crystallography. Antimicrob Agents Chemother. 2007;51:3915. CrossRef
60. Dias MV, Faim LM, Vasconcelos IB, de Oliveira, JS, Basso LA, Santos DS, et al. Effects of the magnesium and chloride ions and shikimate on the structure of shikimate kinase from Mycobacterium tuberculosis. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2007;63:1–6. CrossRef
61. Wehenkel A, Fernandez P, Bellinzoni M, Catherinot V, Barilone N, Labesse G, et al. The structure of PknB in complex with mitoxantrone, an ATP-competitive inhibitor, suggests a mode of protein kinase regulation in mycobacteria. FEBS Lett. 2006;580:3018–22. CrossRef
62. Lombana TN, Echols N, Good MC, Thomsen ND, Ng HL, Greenstein AE, et al. Allosteric activation mechanism of the Mycobacterium tuberculosis receptor Ser/Thr protein kinase, PknB. Structure 2010;18:1667–77. CrossRef
63. He X, Alian A, Stroud RM, Ortiz de Montellano PR. Pyrrolidine carboxamides as a novel class of inhibitors of enoyl acyl carrier protein reductase from Mycobacterium tuberculosis. J Med Chem. 2006;49:6308–23. CrossRef
64. Katritzky AR, Lobanov VS, Karelson M. QSPR: the correlation and quantitative prediction of chemical and physical properties from structure. Chem Soc Rev. 1995;24:279–87. CrossRef
65. Proft FD, Geerlings P. Calculation of ionization energies, electron affinities, electronegativities, and hardnesses using density functional methods. J Chem Phys. 1997;106:3270–9. CrossRef
66. Wadehra A, Ghosh SK. A density functional theory-based chemical potential equalisation approach to molecular polarizability. J Chem Sci. 2005;117:401–9. CrossRef
67. Sabin JR, Trickey SB, Apell SP, Oddershede J. Molecular shape, capacitance, and chemical hardness. Int J Quantum Chem. 2000;77:358–66. CrossRef
68. Moulishankar A, Thirugnanasambandam S. Quantitative structure activity relationship (QSAR) modeling study of some novel thiazolidine 4-one derivatives as potent anti-tubercular agents. J Recept Signal Transduct. 2023;43(3):83–92. CrossRef
69. Valencia J, Rubio V, Puerto G, Vasquez L, Bernal A, Mora JR, et al. QSAR studies, molecular docking, molecular dynamics, synthesis, and biological evaluation of novel quinolinone-based thiosemicarbazones against Mycobacterium tuberculosis. Antibiotics 2022;12(1):61. CrossRef
70. Pietra D, Imbriani M, Borghini A, Giorgi I, Settimo FD, Breschi MC, et al. Structure–activity relationships on purine and 2,3-dihydropurine derivatives as antitubercular agents: a data mining approach. Chem Biol Drug Des. 2011;78:718–24. CrossRef
71. Shagufta, Kumar A, Panda G, Siddiqi MI. CoMFA and CoMSIA 3D-QSAR analysis of diaryloxy-methano-phenanthrene derivatives as anti-tubercular agents. J Mol Model. 2007;13:99–109. CrossRef