INTRODUCTION
Cancer continues to defy the medical science industry with its continued evasion of the current therapeutic agents. The main reason for cancer defiance is that each cancer type is caused by different factors and within the same cancer; different individuals are due to different causes. So, taking all those into consideration, each case of cancer will require specific therapy. Cancer evade therapeutic agents due to its ability to avoid human immune system targeting, replicate radically and metastasis aggressively making current treatments mostly effective in prolonging patients’ life with more chances of relapse [1]. Unrestricted proliferation, self-sufficiency in growth signals, unresponsiveness to inhibitory signals, persistent angiogenesis, evasion from apoptosis, tissue invasion, and metastasis are among the main characteristics found in cancer cells [2]. An estimated 9.7 million people died from cancer, and 20 million new cases were reported in 2022 [3]. Additionally, it was projected that 53.5 million people survived 5 years after receiving a cancer diagnosis, and the estimated 20 million new cases of cancer in 2022 are expected to rise by 77% to over 35 million cases in 2050 [3]. Although South Africa has a high cancer incidence rate, assessing and comprehending its true cancer prevalence is made more difficult by the absence of current and reliable cancer data (the latest NCR report, that is, publicly accessible is from 2017).
To combat the rising cancer cases and advance the Sustainable Development Goals of the United Nations (UN), South Africa has implemented several cancer control programs such as the National Cancer Strategic Framework (2017–2022) which has been created at the level of national policy [4]. Priorities in the framework include lung, colorectal, cervical, prostate, and breast cancers [4]. Prostate cancer ranks as the second highest mortality across all ages and both genders in South Africa [4]. Prostate cancer cells have the potential to metastasize from the prostate to other regions of the body, including the lymph nodes and bones, if untreated, leading to the development of secondary tumors [1]. Annually, new prostate cancer cases are diagnosed in more than 4,300 South African men [1]. On the other hand, pancreatic cancer is ranked the 7th most prevalent cause of cancer-related deaths in both genders and the 12th most prominent cancer worldwide and in South Africa [5]. Although genetic or epigenetic changes are typically the primary cause of cancer, the disease progresses through several phases that are linked to intricate interactions between the tumor cells and their surroundings [6]. The following risk factors have been identified as main contributors to cancer development: age, tobacco smoking, excessive exposure to radiation, poor diet, genetics, sedentary lifestyle, and exposure to carcinogenic chemicals [7]. With current therapeutic agents failing to bring much needed relief to patients and with side effects that remain unbearable to cancer patients, new therapeutic agents are needed for the management and treatments of cancer as a disease [7]. In the search for more effective and affordable therapeutic agents, medicinal plants have become the most reliant option as they have been used by mankind for centuries in the treatment of several illnesses including cancer [8]. To boost efficacy and reduce the side effects of current therapies, more studies are being carried out in combination with medicinal plants and immunotherapy [8]. Many medicinal plants have been shown to inhibit the growth of malignancies and are extensively used because they are affordable, easily accessible, and have little to no side effects [9]. Despite the benefits, some people are worried about the toxicity and safety of herbal and medicinal plants [9]. This study investigates the medicinal plant Leptospermum petersonii (LP) extracts for its potential to manage cancer cases because of its antioxidant, antibacterial, and antifungal properties.
MATERIALS AND METHODS
Crude plant extraction
The leaves of L. petersonii were pre-harvested and collected from KwaZulu-Natal province, South Africa, by Prof. L.R Motadi. Using a mortar and pestle, dried leaves of L. petersonii were selected and ground into a fine powder using a blender. Using a plant-to-extract ratio of 1:10, approximately 80.05 g (methanol solvent: Rochelle chemicals, South Africa) and 80.53 g (ethyl acetate: Sigma-Aldrich, Germany) of crushed leaves were soaked with 800 ml of their respective solvents for approximately 46 and 47 hours, respectively. The Heidolph Rotavac Valve Tec Vacuum Pump was used to filter the plant liquid extracts from the plant debris. The rotary evaporator (MRC, Switzerland) was used in the removal of solvents from samples through evaporation. A laboratory fume hood was used to dry crude extract. Using a mortar and pestling the crude extracts were ground to the powder form of which approximately 5.18 g of methanol and 4.04 g of ethyl acetate crude L. petersonii extracts were retrieved.
Thin-layer chromatography
Aluminum sheets and silica gel-layered TLC plates (UJ chemical store, South Africa) were cut into small 8X8 cm sheets. A 1:10 ratio was used in the preparation of test samples of which approximately 1 g of each sample (EtOAc plant leaves: 1.00 g, methanol plant leaves: 1.01 g, EtOAc crude extract: 1.01 g, and methanol crude extract: 1.03 g) was dissolved with 10 ml of their respective solvent and left overnight to allow for the extraction of compounds. Plates consisting of sample spots were allowed to dry before placing into the TLC developing chamber. The 100% hexane (20 ml) which served as the mobile phase (solvent) was used to run the samples followed by solvent 1:19 ethyl acetate: hexane which equates to 5% ethyl acetate followed by 2:18%–10% EtOAc, 4:16%–20% EtOAc,6:14%–30% EtOAc, 8:12%–40% EtOAc, 10:10%–50% EtOAc solvent ethyl acetate: hexane. Plates were allowed to run for approximately 20–25 minutes until approximately 1 cm from the end of the plate and removed the plate quickly sketched a line over the solvent front. Holding the edge of the plate with a tweezer, the plates were independently dipped into 15% sulfuric acid reagent and heated to stain the spots and visualization of the migration of the test samples.
Column chromatography
The slurry method was used as about one-third of the column was filled with 10% EtOAc in hexane. The amount of silica gel 60 (0.040–0.063 mm) was determined by using an approximate ratio (1:50) of sample (g) /silica (g). An estimated 83.01 g of silica gel was weighed in a beaker and mixed with a solvent (hexane) using a glass rod to make the slurry. The dry crude EtOAc plant extract was loaded into the column (approximately 1.66 g of EtOAc crude extract). Approximately 3 mm of sea sand was added above the crude extract to level and settle the crude uniformly. The compounds were separated from the mixture in this manner. To begin collecting the different fractions in the polytop glass vials, 100 ml of n-hexane (100% solvent) was used to elute the column. TLC was used for the observation of compounds eluting from the column, and the solvent polarity was gradually increased (5%, 10%, 12%, 15%, 17%, 20%, and 25%) until the observation of single bands (pure compound) on TLC plates. A total of 150 fractions of 100 ml each were collected and combined according to their TLC profiles. The distinct chemicals gathered as fractions were subjected to additional analysis (TLC) to clarify their purity of which fractions LP_23 (mixture of fractions LP 19–23) obtained at 10% ethyl acetate in hexane, and fraction LP_39 (mixture of fractions LP 36–39) obtained at 12% ethyl acetate in hexane were obtained and showed possible pure compounds.
Nuclear magnetic resonance chromatography (NMR)
Extracted compounds were dissolved with 0.4 ml of chloroform-d (Sigma Aldrich, Germany) and injected into the NMR glass tube for evaluation. Bruker depth gauge was used to set sample depth (tube’s liquid above the middle bar; approximately 3/5/8 mm). The samples were added to one of the autosampler’s 24 locations and deposited into the sample queue by the Bruker 400 NMR spectrometer, which operates in an automated manner. The autosampler then placed the sample in the queue, inserted it into the NMR magnet, and executed all the NMR experiments (1D ¹H and ¹³C) that were selected.
AlamarBlue assay
Cell culture and maintenance procedure
Dulbecco’s Modified Eagle Medium (DMEM) (ThermoFisher Scientific, USA) was used to culture non-cancerous MRC-5 cell line, PC3 prostate cancer cell line, and pancreatic cancer cell line (MIA PaCa-2 cells) in cell culturing flasks. Under controlled laboratory conditions, cells were incubated with specifications of 95% humidity at temperatures of 37°C and 5% carbon dioxide (CO2). The sub-culturing procedure was conducted until 90% of P3 generation cells.
Seeding and treatment procedure
In the seeding procedure, approximately 1 ml of the cell pellet was thoroughly mixed with 9 ml of supplemented DMEM media in a sterile reagent reservoir. Upon completion of seeding the cells, the cells were incubated with specifications of 95% humidity at temperatures of 37°C and 5% CO2 for 24 hours. The light microscope was used to check for confluency of the cells after the 24-hour incubation period. Upon observation of confluent cells (>90%), cells were treated with different concentrations of the plant extracts (L. petersonii). Volumes of 1 ml treatments were made using the direct dilution of concentrations of 100, 80, 70, 50, and 25 µg/ml in triplicate of which 100 µl was used to treat the cells. The controls of the experiment are as follows: 1 µl into 999 µl of media (0.1% DMSO–negative), 10 µl into 990 µl (1 % methylglyoxal–positive), and 1 ml of supplemented DMEM media (untreated cells). The 96-well plate was incubated with specifications of 95% humidity at temperatures of 37°C and 5% CO2 for 22 hours. The 96-well plate was removed from the incubator after 22 hours and treated with AB (ensuring that AB was not exposed to light) by adding 5 µl of AlamarBlue (AB-ThermoFisher Scientific, USA) to all the respective wells. After adding AB into the wells, the 96-well plate was covered with foil and incubated for 2 hours. The Bio-Tek Synergy HT Microplate Reader was used to read the 96-well plate with the specification: detection mode: fluorescence; excitation: 530/25 nm; emission: 590/35 nm; and sensitivity: 45 mm. Quantification and analysis of results for the cell viability percentage were conducted using Microsoft Excel 2016 with the formula:
GraphPad Prism 8.4.3 (686) was used to generate one-way ANOVA using Dunnett’s comparison test for analysis of cellular viability and graphical plotting of the obtained triplicate results in a bar graph.
Wound healing
An 8-well tissue culture plate was used to seed the cells at a density that after 24 hours, 90% of confluent cells were observed under the microscope. Using a p200 pipette tip, the monolayer across the well’s center was carefully and gently scratched. Scratching was followed by two gentle washes with 2 ml PBS to remove any unattached or dead cells. A 2 ml of treatment solutions were made (untreated cells: DMEM media, 0.1% DMSO, 1% methylglyoxal, and LP-methanolic extract) and pipetted into their labeled wells, respectively. The light microscope was used to observe the cells, and pictures were taken manually (using a phone). While capturing photographs (time: 0, 24, and 38 hours) of the microscope from various angles, the same configurations were used while ensuring to approximately observe the same gap point.
Hoechst staining
A flask of P3 generation of MIA PaCa-2 cells was used in the seeding procedure. Hoechst Dye (ThermoFisher Scientific, USA) was prepared using a 1:2000 ratio. After a 24-hour treatment period, cells were detached from the petri dishes and centrifuged at 2,200 rpm for 2.5 minutes using a tabletop centrifuge. The supernatant was discarded and the washing procedure (3X) was followed using PBS and centrifuged at 2200 rpm for 2.5 minutes. A 50 µl of Hoechst dye was added to each labeled treated cells and foiled and incubated with specifications of 95% humidity at temperatures of 37°C and 5% CO2 for 10 minutes. A 25 µl of cells were fixed on slide and cover slip on top. Cells were then viewed using a fluorescence microscope at 20× and 40× magnification with excitations/emission of 350/461 nm.
ATP assay
In this procedure, cells were seeded and upon observation of 96% confluent cells, old media was discarded, and 2 ml of treatments solutions were made and used to treat the cells for 24 hours. After a 24-hour treatment period, cells were detached and centrifuged at 2,200 rpm for 2.5 minutes using a tabletop centrifuge. The supernatant was discarded, and the washing procedure (2X) was followed using PBS and centrifuged at 2,200 rpm for 2.5 minutes. The supernatant was discarded, 50 µl of fresh media was added to each respective pellet, and approximately 25 µl of cells were added to 50 µl of ATP detection reagent (Promega, USA). Cells were incubated with specifications of 95% humidity at temperatures of 37°C and 5% CO2 for 30 minutes. The Bio-Tek Synergy HT Microplate Reader was used to read the plate with the specification: detection mode: luminescence; excitation: 530/25 nm; emission: 590/35 nm, and sensitivity: 45 mm.
Caspases-Glo 3/7 assay
In this procedure, MIA PaCa-2 cells were used in the seeding procedure, and upon observation of 92% confluent cells, 2 ml of treatments solutions were made (untreated cells: DMEM media, 0.1% DMSO, 1% methylglyoxal, and LP-methanolic extract) to treat the cells. After a 24-hour treatment period, cells were detached from the petri dishes. A 100 µl of Caspase-Glo 3/7 assay reagent (Promega, USA) was added to each well of blank (DMEM media), negative control cells (0.1% DMSO), positive control (1% methylglyoxal) LP-methanolic IC50, and untreated cells in culture medium in their respective wells. The 96-well plates containing treated cells were gently mixed using the plate shaker at 300–500 rpm for 15 minutes. After the 15-minute duration, the Bio-Tek Synergy HT Microplate Reader was used to read the 96-well plate with the specification: detection mode: luminescence; excitation: 530/25 nm; emission: 590/35 nm, and sensitivity: 45 mm.
RNA isolation
Cells were plated overnight and, upon observation of confluency, were treated the next day with the different treatments (DMSO, untreated cells, LP-methanol crude extract, and methylglyoxal ). Cells were incubated for 24 hours with different treatments; cells were detached afterward using 100 µl of trypsin. Using the detached cells, Roche High Pure RNA Tissue Kit (12033674001) (Sigma-Aldrich, Germany) was used in the experiment, and the protocol was conducted according to the manufacturer instruction manual for RNA isolation.
cDNA synthesis and qPCR procedure
SYBR Green fluorescence dye (Bio-Rad, USA) for real-time PCR was used in the amplification of cDNA, and the standard protocol was followed using the instruction manual. The samples were inserted into the CFX connect real-time detection system (Bio-Rad, USA) and thermal cycler instrument to run real-time PCR. Amplification of cDNA was set at 95 °C for 1 minute, followed by 95 °C for 15 seconds and then annealing at 55 °C for 10 seconds and lastly extension and final extension at 55 °C for 10 seconds and 55 °C for 10 seconds, respectively. Quantification of amplified cDNA for determination of relative gene expression was conducted using Microsoft Excel 2016 with the delta-delta Ct 2–??Ct method relative to the β-actin housekeeping gene. GraphPad Prism 8.4.3 (686) was used to generate bar graphs for analysis of gene expression. Table 1 shows the primer sequences used in the study for analysis of gene expression.
Table 1. Primer sequences of analyzed regulatory genes.
| Gene | Primer sequences |
|---|---|
| BCL-2 | Primer 1: 5′-GCTATAACTGGAGAGTGCTGAA -3′ Primer 2: 5′-TGTTAATATCAGTCTACTTCCTCTGTG -3′ |
| p53 | Primer 1: 5′-GACACGCTTCCCTGGATTG-3′ Primer 2: 5′-GACGCTAGGATCTGACTGC -3′ |
| TNF-α | Primer 1: 5′-CCGAGGCAGTCAGATCATCTT-3′ Primer 2: 5′-AGCTGCCCCTCAGCTTGA-3′ |
| RB-1 | Primer 1: 5′-AGCTCTCCAGGTGAACTTGC-3′ Primer 2: 5′-TTGCTCTGCTGTTGACAGGT-3′ |
| CHEK 1 | Primer 1: 5′-AGCCACCATGAAGGAGTTTG-3′ Primer 2: 5′-TGTTGTTGGCTTTTCCAGGA-3′ |
| FAS | Primer 1: 5′-CAGAACTTGGAAGGCCTGCATC-3′ Primer 2: 5′-TCTGTTCTGCTGTGTCTTGGAC-3′ |
| WEE 1 | Primer 1: 5′-GGAGTGTTGGGGAAGGTTTC-3′ Primer 2: 5′-CTTCAGCCCATTTGGTCTTG-3′ |
| AKT | Primer 1: 5′-TCTATGGCGCTGAGATTGTG-3′ Primer 2: 5′-CTTAATGTGCCCGTCCTTGT-3′ |
| β-actin | Primer 1: 5′-GTCTTCCCCTCCATCGTG-3′ Primer 2: 5′-GATGCCGTGCTCGATGG-3′ |
DNA fragmentation analysis
The Zymo Research Quick DNA Miniprep Plus Kit, Lot No: 240057 (Zymo Research, USA) was used in the DNA extraction process in accordance with the manufacturer’s instructions. Agarose gel electrophoresis (LonzaSeaKem LE Agarose Lot No: 0000313053, Size 500 g: Fisher Scientific, USA) was then used for analysis of DNA fragmentation. GelDoc Go, Imaging System 12009077 (Bio-Rad, USA), was used to obtain image visualization of band migration on the agarose gel.
RESULTS
TLC and column chromatography illustration of compound isolation
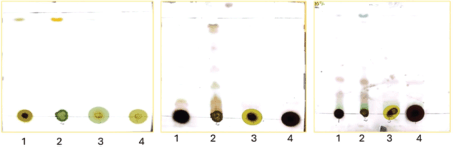
The results of the TLC and column chromatography are presented in this section. The purpose was to identify the different chemicals present in the extract. Fifteen percentage aqueous sulfuric acid (acid staining method) and heat were used to develop the spots. As shown later in our TLC plates, there were fewer compounds identifiable in hexane: ethyl acetate separation 100%, 5%, and 10% as compared to the 20%, 30%, 40%, and 50%, respectively (Figs. 1 and 2, Table 2). Samples labeled 1 and 2 are plant crude extracts, while samples labeled 3 and 4 are plant leaves soaked in the respective solvents. It was observable with the TLC plate bands that LP crude extract showed better compound extraction as compared to LP leaves soaked in solvent (samples 3 and 4) (Fig. 3).
 | Figure 1. TLC plates illustration of the compound separation at different solvent systems (hexane: ethyl acetate) percentage separation (100%, 5%, and 10% respectively). [Click here to view] |
 | Figure 2. TLC plates illustration of the compound separation at different solvent system (hexane: ethyl acetate) percentage separation (20%, 30%, 40%, and 50%, respectively). [Click here to view] |
Table 2. 1H and 13C NMR DATA of compound 1.
| Position | 1H (nH, m, J(Hz)) | 13C |
|---|---|---|
| 1 and 38 | 0.85 (6 H, t, J=7.01) | 14.1 |
| 2 and 37 | 1.54 (4H, Brs) | 32.0 |
| 3 and 36 | 1.23 (4H, m) | 22.7 |
| 4–35 | 1.11–1.40 (64H, Brs) | 29.6–29.8 |
 | Figure 3. TLC plate observation of compounds LP_23, LP_39, and LP_52. [Click here to view] |
At 20% and 30%, sample 2 (not clearly visible due to inadequate image resolution development), 5 distinct bands (Fig. 2) were measured and Rf (retention factor) values were determined. At 20% and 30% hexane: ethyl acetate separation (sample 2: LP ethyl acetate crude extract) distinct band separation was observed with Rf values of 0.96, 0.94, 0.90, 0.69, and 0.52. Rf values can vary depending on the experimental conditions, solvent system, stationary phase, and type of compound [10]. Nonetheless, calculated Rf values are indicative of alkane, aromatic, terpenes, and alkaloids.
1–LP methanol crude extract
2–LP ethyl acetate crude extract
3–LP leaves ethyl acetate crude extract
4–LP leaves methanol crude extract
Due to time constraints and limited resources, column chromatography was conducted with LP ethyl acetate crude extract due to its adequate compound separation solvent system and availability of crude extract. Approximately 150 polytop vial fractions were collected. TLC analysis was conducted, and fractions with similar single bands were combined (increased concentration of compound). Fractions LP_23 (mixture of fractions LP 19–23) obtained at 10% ethyl acetate in hexane, LP_39 (mixture of fractions LP 36–39) obtained at 12% ethyl acetate in hexane, and LP_52 (mixture of fractions LP 52–58) obtained at 15% ethyl acetate in hexane were identified as pure compounds and further analyzed for their purity was conducted.
Characterization of the isolated compounds compound 1 (LP 23)
Compound 1 was isolated as a white powder using a mixture of hexane/EtOAc (ratio), it was then purified through recrystallization for further studies. Its proton NMR spectrum (500 MHz, CDCl3) displayed three sets of peaks in a strong field attributable to aliphatic protons. Among those peaks, one appeared at δH 0.85 (m, J= 7.01 Hz, 6H) attributable to two terminal methyl groups in an aliphatic chain; a broad singlet at δH 1.22–1.40 (Brs) attributable to methylene (-CH2-) proton in a long aliphatic chain. A third peak at δH 1.54 (Brs) was attributed to the methylene (-CH2-) protons in the alpha position of our methyl. On its carbon NMR spectrum (126 MHz, CDCl3), we observed an intense peak at δC 29.7 corresponding to the carbon peaks of the aliphatic chain observed on proton NMR. Three other aliphatic carbons were observed at δC 14.1, 32.0, and 22.7 attributable to the terminal methyl, the methylene at position alpha to the methyl, and the methylene in beta position to our methyl, respectively. All this information coupled with that from the literature enabled us to deduce that this compound is an alkane (hydrocarbon) called octatriacontane [11,12,13].
Characterization of compound 2 (LP 39)
Compound 2 was obtained as a white needle-like crystal. The ESI-MS (positive mode) showed a molecular ion peak at m/z 285.1619, corresponding to the molecular formula C19H39OH. The 13C NMR spectrum showed nine signals, of which signals at δC14.2 and 63.0 were characteristic of the presence of a terminal methyl (-CH3) and an oxymethylene (-CH2OH), respectively. The remaining signals resonating at δC 29.3–29.6 were assigned to the other aliphatic -CH2- groups of the molecule, suggesting that compound 2 to be aliphatic straight-chain primary alcohol. The 1H NMR spectrum of compound 2 revealed a downfield signal at δH 3.55 (2H, t, J=7.3Hz) which is unambiguously assigned to the oxymethylene proton -CH2OH group. The intense broad signal at δH 1.14–1.26 (34H, m) was assigned to the aliphatic proton of the long chain on our compound. Based on the above data, together with those in the literature, compound 2 was identified as a fatty alcohol called 1-nonadecanol11,12,13.
Evaluation of cytotoxic effect of methanol and ethyl acetate crude extracts of L. petersonii using cellular viability test: AlamarBlue assay
The cytotoxic effects of L. petersonii methanolic and ethyl acetate extracts at various concentrations on pancreatic (MIA PaCa-2) and prostate cancer (PC3) cell lines including MRC5 normal cell line were measured. The crude extracts were diluted at the concentrations of 100 (1 µl of crude extract into 999 µl of media), 80 (0.8 µl in 999.2 µl), 70 (0.7 µl in 999.3 µl), 50 (0.5 µl in 999.5 µl), 25 (0.25 µl in 999.75 µl), and 12 µg/ml (0.12 µl in 999.88 µl) to make 1 ml treatments using direct dilution of concentrations of which 100 µl of the treatment was used to treat the cells. The results demonstrate a concentration-dependent effect on MIA PaCa-2 and PC3 cancer cell lines following treatment with methanolic crude extract, with the maximum concentration of 100 µg/ml exhibiting the highest inhibitory impact of 48.54% on MIA PaCa-2 cells but less inhibitory in PC3 of 22%, as shown in Figures 6 and 8 The EtOAc crude extract has shown little toxicity in all cell lines as shown in Table 7. Although at high concentrations (Fig. 8), there is significant inhibition of cellular viability of MIA PaCa-2 cells. The MRC5 cell line was the least to show any inhibitory growth by both extracts with a predictable 250 µg/ml IC50 (Table 7) of the cells. The cell line PC3 had a tolerable IC50 of 184.91 µg/ml (methanolic crude extract). From the results observed, the workable concentration that was able to inhibit 50% of cell growth was 101.595 µg/ml (methanolic crude extract) in MIA PaCa-2 cell lines. The other concentration and cell line were not desirable for continuation as they did not meet the standard for plant concentration desired for continuation in vivo studies. These results suggested that the crude extract was also cell-type-dependent. For this reason, only the MIA PaCa-2 cell line with a concentration of 101.595 µg/ml was continued with the other experiment mentioned later. Figure 6–8 are the illustrations of quantified values calculated using GraphPad Prism 8.4.3 (686) for the 3 tested cell lines showing the mean value difference with untreated cells and the p value (Table 4–6).
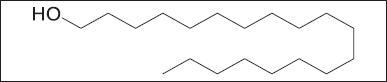 | Figure 5. Chemical structure of compound 2. [Click here to view |
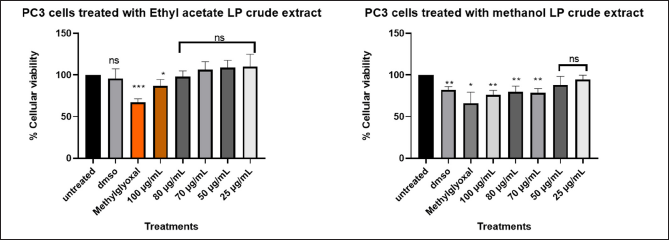 | Figure 6. Graphical illustration of the cellular viability of PC3 cells. [Click here to view] |
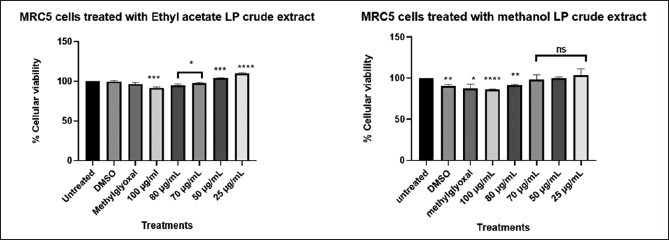 | Figure 7. Graphical illustration of the cellular viability of MRC5 cells. [Click here to view] |
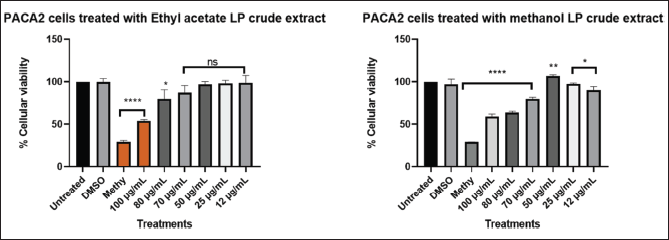 | Figure 8. Graphical illustration of the cellular viability of MIA PaCa-2 cells. [Click here to view] |
Different concentration of direct dilution ranging from 100 µg/ml to 25 µg/ml (Fig. 6) of LP-EtOAc and methanolic crude extract was used to test its antiproliferative effect. Cells seeded with DMEM media represented untreated cells, the negative control group was cells treated with 100 µl of 0.1% DMSO, while the positive control group were cells treated with 100 µl of 1% methylglyoxal. The Asterisk star (*) observed above the bar graph indicates the significant difference in the t-test conducted using GraphPad Prism 8.4.3 (686) between untreated cells and the different treatments. NS indicated a non-significant difference between the treated and untreated cells.
Different concentration of direct dilution ranging from 100 µg/ml to 25 µg/ml (Fig. 7) of LP-EtOAc and methanolic crude extract was used to test its antiproliferative effect. Cells seeded with DMEM media represented untreated cells, the negative control group was cells treated with 100 µl of 0.1% DMSO, while the positive control group were cells treated with 100 µl of 1% methylglyoxal. The Asterisk star (*) observed above bar graph indicates the significant difference in the t-test conducted using GraphPad Prism 8.4.3 (686) between untreated cells and the different treatments. NS indicated a non-significant difference between the treated and untreated cells.
Different concentration of direct dilution ranging from 100 µg/ml to 25 µg/ml (Fig. 8) of LP-EtOAc and methanolic crude extract was used to test its antiproliferative effect. Cells seeded with DMEM media represented untreated cells, the negative control group was cells treated with 100 µl of 0.1% DMSO, while the positive control group were cells treated with 100 µl of 1% methylglyoxal. The Asterisk star (*) observed above the bar graph indicates the significant difference in the t-test conducted using GraphPad Prism 8.4.3 (686) between untreated cells and the different treatments. NS indicated a non-significant difference between the treated and untreated cells.
Wound healing image illustration of the cellular migration and morphological changes cancerous cells undergo upon treatment with different experimental reagents
Using the wound healing test, which monitors the cell’s capacity to metastasize to different areas of the body, MIA PaCa-2 was examined at a concentration of 101 µg/ml. Cell lines were treated with 0.1% DMSO, 1% methylglyoxal, and IC50 of the crude extract after an artificial wound was made in the cell monolayer. Column 1 of Figure 9 represents untreated cells, column 21% methylglyoxal, column 30.1% DMSO, and column 4–IC50 of crude methanolic extract. It can be noted that MIA PaCa-2 (Fig. 9–C1) untreated and DMSO-treated cells (Fig. 9–C3) effectively moved into the artificial wound and formed an epithelial monolayer within 30 hours. While methyl-treated cells (Fig. 9–C2) did not migrate at all, and IC50-treated cells (Fig. 9–C4) were unable to close the gap and died. Based on these findings, it is possible to deduce that L. petersonii can prevent MIA PaCa-2 cancer cells from migrating, implying that the plant extract is capable of preventing metastasis.
Graphical illustration of Caspase 3/7 activity induced by IC50 of LP-methanolic extract on MIA PaCa-2 cells with one-way ANOVA using Dunnett’s comparison test for analysis of Caspase 3/7 activity
Given that methanolic crude extract was effective against MIA PaCa-2 cancer cells, it was crucial to determine the mechanism of cell death induced by L. petersonii. To do this, the Caspase-Glo 3/7 assay was used to investigate apoptosis induction in the cells. Relative Luminescence Units (RLUs) were used to assess caspase activity. As can be observed in Figure 10, the obtained caspase levels for MIA PaCa-2 were less than 5000 RLU in untreated and DMSO-treated cells, while in the crude extract was shown to have increased to over 9000 RLU and in methylglyoxal was over 18000 RLU. In comparison to untreated and DMSO-treated cells, the IC50 crude methanolic extract has shown a 1.8-fold increase. There is a 2.0-fold increase from 9000 RLU (IC50 crude methanolic extract) to 18000 RLU (methylglyoxal). An increase in RLU indicates a significant rise in caspase activity. Apoptosis induction in the cells is shown by an increase in caspase activity levels as they are mainly the initiators and executors. So, an increase in caspase activity assay is an indication of perhaps apoptosis induction. The Bio-Tek Synergy HT Microplate Reader was used in the detection of Caspase 3/7 luminescence by setting the instrument to read the plate with the specification: detection mode: luminescence; excitation: 530/25 nm; emission: 590/35 nm, and sensitivity: 45 mm.
Graphical illustration of ATP detection assay induced by IC50 of LP-methanolic extract on MIA PaCa-2 cells with one-way ANOVA using Dunnett’s comparison test for analysis of Caspase 3/7 activity
Cells can be differentiated between live and dead cells based on their ATP levels. RLUs are used to quantify the amount of ATP in MIA PaCa-2 cancer cells following treatment with L. petersonii crude methanolic extract IC50. ATP levels measured for MIA PaCa-2 were significantly reduced in both methylglyoxal and crude extract from the untreated 13000RLU to 3400 and 4600RLU, respectively. This shows a significant 3.8- to 2.8-fold RLU fold decrease (Fig. 11). According to the literature, when cells undergo cell death, their ATP levels plummet significantly in comparison to live cells. The Bio-Tek Synergy HT Microplate Reader was used in the detection of ATP luminescence by setting the instrument to read the plate with specification: detection mode: luminescence; excitation: 530/25 nm; emission: 590/35 nm, and sensitivity–45 mm.
Hoechst staining analysis of cellular death induced by LP plant extract
The integrity of the cell nucleus may be examined using the fluorescent dye Hoechst stain, which binds to the nucleus of cells [9]. This test was carried out to further illustrate the apoptotic impact that L. petersonii methanolic crude extract has on MIA PaCa-2 cancer cells, as nuclear fragmentation and condensation is one of the primary characteristics of apoptosis. In terms of MIA PaCa-2 cells following treatment, it showed that untreated cells contain an intact nucleus as indicated by the bright blue color, and this is also apparent in cells treated with 0.1% DMSO. In contrast, the nuclei of cells treated with methylglyoxal and crude extract IC50 have condensed, some of them have disintegrated, and there is a reduction in the number of cells for these treatments. Hoechst staining was conducted to ascertain the mechanism of action of the L. petersonii methanolic crude extract. In comparison to other cytotoxicity tests, Hoechst staining is less toxic and binds to live cells allowing analysis of nuclear structure and dynamics in live cells [14].
Image of DNA fragmentation agarose gel
Since caspase activation suggested potential apoptosis activation, the next objective was to further validate the occurrence of apoptosis by assessing the integrity of the DNA. Under normal circumstances, DNA remains a singular intact component when extracted. However, if apoptosis occurs, caspase-3 and -7 activation triggers the cleavage of cellular components, including DNA. Figure 14 shows the DNA of untreated and treated cells. Untreated and 0.1% DMSO-treated cells revealed one intact band, with no sign of fragmentation. On the other hand, crude L. petersonii-treated cells displayed a distinct series of bands indicating DNA fragmentation. Although methylglyoxal does not show any DNA this might be due to the number of cells that died and little DNA collected from remaining cells that could not show on the gel.
Regulatory gene expressions
To predict the mechanism of action of L. petersonii methanolic crude extract, qPCR was employed to evaluate the expression of both apoptosis-inducing and cell cycle genes following treatment with the IC50 as determined by AlamarBlue. The real-time PCR was performed to evaluate the regulation of the genes TP53, TNF, Bcl-2, CHEK1, RB1, WEE1, FAS, and AKT1 following medicinal plant extract treatment. First, extraction of messenger-RNA (mRNA) was conducted using the RNA cell miniprep kit; this was followed by usage of the GoScript Reverse Transcriptase for reverse transcription procedure of which both procedures were conducted following the manufacturer’s specified protocol. Standard RT-PCR was then performed, and graphical amplifications indicating the expression levels of the different apoptotic and cell cycle genes were observed. It was observed from the results obtained that the following genes: checkpoint 1, RB1, p53, and AKT1 (Figs. 15 and 16) were significantly increased in MIA PaCa-2 cells. While FAS and TNF (Fig. 15) increase was not significant which might suggest their little impact in the pathway initiated by crude extract. Bcl2, which is a negative regulator of apoptosis, was low as compared to untreated cells. In contrast, checkpoint 1, RB1, p53, and ATK1 were not increased, but in some instances, they were decreased as compared to untreated cells.
DISCUSSION
When it comes to the possible pharmacological effects of crude plant extracts, the identification of active components in medicinal plants is crucial and plays a critical role in the phytochemical identification process [15]. For this, thin-layer chromatography (TLC) has the potential to help profile the phytochemicals of any crude extract [15]. In this study, TLC was utilized to determine the system that provides the most effective separation described as the optimal solvent system (mixture of solvents: hexane and EtOAc). The TLC results were analyzed (Figs 1 and 2), and it was determined that 20% of sample 2—LP ethyl acetate crude extract, showed distinct bands that 5 Rf values were calculated. The technique involved trial and error as the different samples were run at different solvent system percentages of hexane: ethyl acetate, it focused on reporting the chromatographic results in each solvent system as to efficiently ascertain which of the mixture’s components will move more quickly as the solvent system’s polarity is changed and which solvent system provides the best separation. The distinct band separation observed with 20% EtOAc crude extract was used to conduct column chromatography. Sulfuric acid was used in spot development due to its versatility in the detection of organic compounds including non-UV fluorescent substances [11]. Sulfuric acid is also inexpensive, not requiring specialized laboratory equipment, relatively spots remain stable over time, and rapid visualization with increased sensitivity [11].
Compounds from a mixture can be purified using column chromatography, which was the technique that was used in the purification process to ascertain compounds from the plant extract. This technique involved a liquid solvent, known as the mobile phase (hexane: ethyl acetate), gently descending a cylinder-shaped glass column filled with silica gel, the stationary phase, from the top with the aid of gravity [12]. Two compounds labeled as LP-23 and LP-39 (Figs 4 and 5) were identified using column chromatography from ethyl acetate crude extract that were further analyzed using NMR chromatography. Because of the lack of resources and time constraints and the fact that the column chromatography method is not automated and necessitates regular monitoring and assessment, methanol crude extract compound extraction was not performed. The use of NMR in natural product research is currently trending toward direct NMR analysis of crude mixtures to gather information on biological activity and molecular structure without the need for laborious separation [12]. We were able to identify the chemicals as an alkane (hydrocarbon) called octatriacontane (compound 1) and a fatty alcohol called 1-nonadecanol (compound 2) using 1H and 13C NMR data (Tables 2 and 3). The small fractions of the compounds (less than 1 g) produced by the small starting plant material (about 1.66 g of EtOAc crude extract), made it challenging for further study using the compounds. Due to the small amount of the compounds and repeated testing and analysis for confirmation, this depleted the compounds and thus were unable to conduct a cytotoxic assessment of the compounds.
Table 3. 1H and 13C NMR DATA of the compound.
| Position | 1H (nH, m, J(Hz)) | 13C |
|---|---|---|
| 1 | 3.55 (2H, t, J=6.65) | 63.0 |
| 2 | 1.58 (2H, q, J=6.88) | 32.8 |
| 3 | 1.63 (2H, m) | 25.7 |
| 4–16 | 1.14–1.26 (34H, m) | 29.3–29.6 |
| 17 | 1.25 (2H, m) | 31.9 |
| 18 | 1.25 (2H, m) | 22.6 |
| 19 | 0.90 (3H, t, J= 6.86) | 14.0 |
According to the principle of similarity and inter-miscibility (like dissolves), solvents whose polarity values are close to those of the solutes tend to react better, and vice versa [13]. As a result of its high polarity, methanol solvent is often utilized and highly recommended due to the substantial high extraction yields [13]. Furthermore, many organic molecules found in plants are among the many polar and moderately polar substances that methanol may dissolve since it is a polar molecule [6]. Ethyl acetate (EtOAc) is another widely used solvent in the pharmaceutical industry due to its ability to extract active components from therapeutic plants [6]. This solvent interacts well with a variety of botanical compounds, extraction of volatile compounds, specific types of lipids, essential oils, and low boiling point and can be used with other solvents to improve extraction [13]. However, studies have shown the methanol solvent extraction system to yield total more flavonoid, phenolic, and antioxidant activity in comparison to EtOAc [12,14,16]. This study examined the effects of different extracts on the cytotoxic effect of the plant extract using methanol and EtOAc as solvents and it was observed (Table 7) with the cellular viability test that the methanolic crude extract exhibited greater cytotoxic effects than EtOAc crude extract. These findings may be due to the polarity range of methanol solvent as its able to extract polar and semi-polar compounds and consist of hydroxyl group that is able to form hydrogen bonds with compounds such as phenolics, flavonoids, and sugars [13]. The plant L. petersonii is also found to have high phenolic, terpenoid, alkaloid, and polyphenol compound content which are polar compounds making methanol the most suitable extraction solvent [14]. Methanol has also been found to have higher extraction efficiency in comparison to ethyl acetate or hexane (less polar solvents) due to its penetration ability for enhancing the extraction of intracellular compounds [2,11,17]. This could imply methanol crude extract consists of total higher compound contents that synergistically execute an antiproliferative effect.
Table 4. GraphPad Prism 8.4.3 (686) generated one-way ANOVA using Dunnett’s comparison test for analysis of cellular viability of PC3 cells treated with LP-EtOAc and methanolic crude extract with untreated cells as the control group.
| PC3 cells treated with LP-EtOAc crude extract | PC3 cells treated with LP-methanolic crude extract | |||||
|---|---|---|---|---|---|---|
| Dunnett’s comparison test | Mean value for treatments | p value | Mean difference with untreated | Mean value for treatments | p value | Mean difference with untreated |
| Untreated versus DMSO | 95.61 | 0.5482 | −4.394 | 82.18 | 0.0012 | −17.82 |
| Untreated versus Methylglyoxal | 67.03 | 0.0002 | −32.97 | 66.03 | 0.011 | −33.97 |
| Untreated versus 100 µg/ml | 86.91 | 0.0408 | −13.09 | 76.05 | 0.0017 | −23.95 |
| Untreated versus 80 µg/ml | 98.07 | 0.6521 | −1.93 | 79.58 | 0.0072 | −20.42 |
| Untreated versus 70 µg/ml | 106.1 | 0.3349 | 6.132 | 78.59 | 0.0018 | −21.41 |
| Untreated versus 50 µg/ml | 108.9 | 0.1528 | 8.885 | 87.95 | 0.115 | −12.05 |
| Untreated versus 25 µg/ml | 110.1 | 0.3056 | 10.05 | 94.59 | 0.1411 | −5.409 |
Table 5. GraphPad Prism 8.4.3 (686) generated one-way ANOVA using Dunnett’s comparison test for analysis of cellular viability of MRC5 cells (normal cells) treated with LP-EtOAc and methanolic crude extract with untreated cells as the control group.
| MRC5 cells treated with LP-EtOAc crude extract | MRC5 cells treated with LP-methanolic crude extract | |||||
|---|---|---|---|---|---|---|
| Dunnett’s comparison test | Mean value for treatments | p value | Mean difference with untreated | Mean value for treatments | p value | Mean difference with untreated |
| Untreated versus DMSO | 99.71 | 0.6618 | −0.2928 | 90.2 | 0.002 | −9.796 |
| Untreated versus methylglyoxal | 96.44 | 0.051 | −3.556 | 87.78 | 0.0149 | −12.22 |
| Untreated versus 100 µg/ml | 91.6 | 0.0002 | −8.397 | 86.63 | <0.0001 | −13.37 |
| Untreated versus 80 µg/ml | 94.88 | 0.0108 | −5.116 | 91.6 | 0.0002 | −8.397 |
| Untreated versus 70 µg/ml | 97.63 | 0.014 | −2.373 | 98.05 | 0.6052 | −1.952 |
| Untreated versus 50 µg/ml | 103.8 | 0.0007 | 3.781 | 99.91 | 0.933 | −0.09215 |
| Untreated versus 25 µg/ml | 109.9 | <0.0001 | 9.867 | 103.9 | 0.4078 | 3.93 |
Table 6. GraphPad Prism 8.4.3 (686) generated one-way ANOVA using Dunnett’s comparison test for analysis of cellular viability of MIA PaCa-2 cells treated with LP-EtOAc and methanolic crude extract with untreated cells as the control group.
| MIA PaCa-2 cells treated with LP-EtOAc crude extract | MIA PaCa-2 cells treated with LP-methanolic crude extract | |||||
|---|---|---|---|---|---|---|
| Dunnett’s comparison test | Mean value for treatments | p value | Mean difference with untreated | Mean value for treatments | p value | Mean difference with untreated |
| Untreated versus DMSO | 100 | 0.9866 | 0.03887 | 96.84 | 0.4275 | −3.165 |
| Untreated versus methylglyoxal | 28.9 | <0.0001 | −71.1 | 28.9 | <0.0001 | −71.1 |
| Untreated versus 100 µg/ml | 53.97 | <0.0001 | −46.03 | 59.1 | <0.0001 | −40.9 |
| Untreated versus 80 µg/ml | 79.91 | 0.0315 | −20.09 | 63.63 | <0.0001 | −36.37 |
| Untreated versus 70 µg/ml | 87.43 | 0.0533 | −12.57 | 79.71 | <0.0001 | −20.29 |
| Untreated versus 50 µg/ml | 97.04 | 0.1911 | −2.958 | 106.8 | 0.0012 | 6.762 |
| Untreated versus 25 µg/ml | 98.31 | 0.4412 | −1.691 | 97.62 | 0.017 | −2.376 |
| Untreated v versus 12 µg/ml | 98.6 | 0.7964 | −1.404 | 90.15 | 0.0148 | −9.85 |
Table 7. Calculated IC50 values of the LP-EtOAc and methanolic crude extract cytotoxic effect on the different cell lines.
| Plant crude extract | MRC5 cell line | PC3 cell line | MIA PaCa-2 cells |
|---|---|---|---|
| LP-Methanolic | 249.505 µg/ml | 184.91 µg/ml | 101.595 µg/ml |
| LP-EtOAc | 259.02 µg/ml | 203.465 µg/ml | 116.975 µg/ml |
Because of its affordability, more sensitive, and non-destructive nature, AlamarBlue (AB) assay was chosen above other reagents such as MTT and Cell-Titer Glo. AB assay consists of quantitative and qualitative analysis of which qualitative analysis involves observation of the color change, which is indicative of metabolically live cells, while quantitative analysis utilizes microplate reader that uses fluorescence or absorbance to quantitatively determine the cellular viability and cytotoxicity of the treatments [18]. It was observed upon the addition of 5 µl of AB each cell well turned blue and after the 2-hour incubation period, some cell wells turned pink, purple, and other remained blue (Fig . 6). In accordance with AB assay principle, the quantity of live cells determines how much fluorescence is generated and in actively functioning cells, the non-fluorescent indicator dye resazurin is chemically reduced to vivid red-pink, fluorescent resorufin [18]. MS Excel was used to quantify the percentage cellular viability and determine the IC50 value of the plant extract. The IC50 (50 % inhibitory concentration) is a crucial metric in in vivo cancer research involving plant extracts since it denotes the concentration at which a plant extract begins to significantly hinder the proliferation of cancer cells [19,20]. Based on the American National Cancer Institute (NCI), crude extracts deemed highly cytotoxic possess IC50 less than (<) 20 μg/ml or 10 μM after 48 hours or 72 hours, promising crude extracts for purification are those with IC50 with an upper limit criterion of 30 μg/ml [20]. The LP-crude extract was shown to be effective at > 100 µg/ml concentrations with methanolic extract showing higher potency (workable IC50 of 101.595 µg/ml) in comparison to EtOAc extract in MIA PaCa-2 cell line. The LP-crude extract showed insignificant cytotoxicity on PC3 cell line as IC50 of 184.91 µg/ml (methanolic extract) and 203.465 µg/ml (EtOAc extract) were obtained. Due to their high values (Table 7) and IC50 concentrations proximity to the IC50 of normal cell line may also be toxic to other normal organs. Therefore, a successful anticancer medication should target cancer cells while maintaining healthy cells [20]. The crude extract was thus tested on normal cell line (MRC5) to ascertain its non-toxicity on healthy cells. The LP-crude extract showed insignificant cytotoxicity (Fig. 12) on MRC5 cell line with IC50 of 249.505 µg/ml (methanolic extract) and 259.02 µg/ml (EtOAc extract). The plant extract showed a considerable cytotoxic effect on MIA PaCa-2 cells (Fig. 12). Although IC50 of 101.595 µg/ml (methanolic extract) is considerably high according to acceptable NCI crude extract IC50 concentrations, other publishing and scientists, however, believe that greater values and a plant extract may be useful as cytotoxic at concentrations as high as ~100 g/ml [21]. With a molecular weight of 300 g/mol, molecules with 10 µM (about 3.4 µg/ml) are thought to be viable candidates for therapeutic development; therefore, an IC50 of 100 µg/ml (about 0.33 µM) would be deemed acceptable [20]. From the experiment, a trend was observed of methanolic extract having higher potency as compared to EtOAc extract. Since methanolic crude extract produced more favorable outcomes than EtOAc extract, this study chose to proceed with it. The control groups in the assay included dimethyl sulfoxide (DMSO)—negative control and methylglyoxal—positive control group. A significant aprotic solvent, DMSO can dissolve a broad range of polar and nonpolar compounds that would otherwise be weakly soluble [19]. This, together with its perceived low toxicity at quantities less than 10%, has made it widely used [19,22,23]. In the procedure, concentration of 0.1% DMSO was used which showed non-toxic traits in all cell lines.
 | Figure 9. Microscopic imaging of wound healing assay at different time points (illustration from Rows 1 to 3–0 hours, 24 hours, and 38 hours) at 10× magnification of MIA PaCa-2 cells treated with 1% methylglyoxal (positive control) wounded with p200 pipette tip. [Click here to view] |
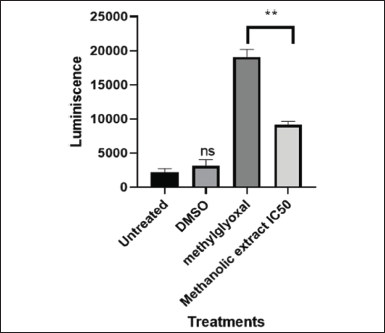 | Figure 10. Graphical illustration of Caspase 3/7 activity induced by IC50 value (101.595 µg/ml) of LP-methanolic extract on MIA PaCa-2 cells. [Click here to view] |
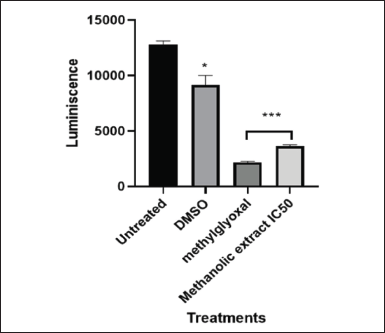 | Figure 11. Graphical illustration of ATP detection induced by IC50 value (101.595 µg/ml) of LP-methanolic extract on MIA PaCa-2 cells. [Click here to view] |
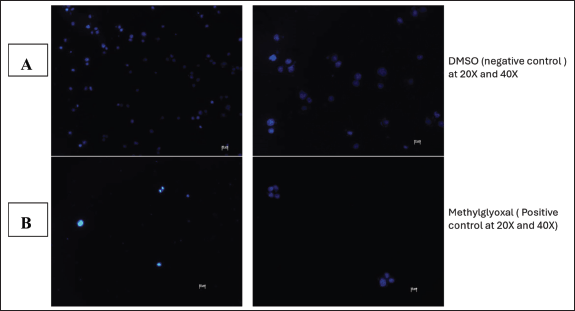 | Figure 12. Image illustration of Hoechst staining of 0.1% DMSO (A) and 1% Methylglyoxal (B) treated MIA PaCa-2 cells at 20X and 40X magnification respectively. [Click here to view] |
Cell migration is simply defined as an essential process that allows the transfer of single cells, cell clusters, and cell sheets to adapt and move into the appropriate location in an environment to perform their task [24]. Sheet migration is the kind of collaborative cell movement that the wound healing assay examines [24]. Monolayers of epithelial and endothelial cells that migrate in two dimensions while preserving their intercellular connections are examples of this migration [25]. Moving cells can, however, become out of control and contribute to a variety of pathological events, including inflammation and cancer metastasis [26]. Cell invasion is the term for the 3D migration of cells through an extracellular matrix (ECM), a process most often connected with the metastasis of cancer cells [24]. Cell invasion plays a crucial role in a wide range of pathological and physiological processes in numerous branches of biology, including inflammation, cancer, wound healing, cell proliferation, and differentiation. The significant roles played by cell migration, invasion, and adhesion in metastasis make it essential to study and comprehend to combat cancer [24]. Furthermore, one of the main causes of cancer-related mortality and morbidity is the spread of cancer cells to peripheral organs and the subsequent elimination of those cells [26].
It was observed over the period of 38 hours that MIA PaCa-2 cells untreated control exhibited aggressive cellular migration (Fig. 9-R3C1) (Row 3 Column 1). Within a 24-hour time lapse, the gap closed, illustrating the aggressive nature of MIA PaCa-2 cells in metastasis. Contributing factors such as smaller cell culturing flask (6-well plate) allowed cells to overcrowd and eventually hindered cell division and health leading to death as observed by change in morphology (epithelial cells–round cells) at 38 hours (Fig. 9-R3C1). Figure 9-C4 illustrates MIA PaCa-2 cells treated with IC50 of methanol crude extract in which the cells seem to maintain their structural integrity (Fig. 9-C4) and showed insignificant migration indicating cytotoxic potency of the plant extract in hindrance of metastasis. The analysis of MIA PaCa-2 cells at 38 hours (Fig. 9-R3C4) showed cell rounding and blebbing, as well as cell shrinkage, which are characteristic indicators of apoptotic cell death. Although the crude extracts may be considered to have apoptotic effects, further testing would be necessary to determine exactly what kind of cellular death they cause due to a lack of cellular migration toward the scratch wound, and the LP-methanol crude extract may be considered to have antimetastatic effect. Figure 9-C3 illustrates cells that have been treated with 0.1% DMSO (negative control). As observed in the image, in Figure 9-R2C3, there is an increase in cellular population (clustering cells) with observation of round and spindle-shaped (typical morphology of MIA PaCa-2 cells) as compared to Figure 9-R1C3, the cellular morphology of MIA PaCa-2 cells then changes after a 38-hour incubation period to more round cells and migrate towards the gap (Fig. 9-R3C3). This could be due to limited space and although DMSO has been scientifically approved and classified in the safety solvent category for research use, some studies have found different concentrations of DMSO may alter the way cells activate in culture, induce lymphocyte activation, and have anti-inflammatory properties [27]. In biomedical and pharmacological research, DMSO is still the preferred solvent despite the possibility of cytotoxicity and physiological interference. To guarantee reliability and precision of results, it is still crucial to comprehend potential interactions and choose the ideal, non-toxic DMSO concentration in each cell model and experimental conditions [17]. A known-effective chemical (methylglyoxal) was used to treat the cells. Figure 9-C2 is an illustration of cells treated with 1% methylglyoxal (positive control). As observed from the images, there was a slight increase in cellular population (Fig. 9-R1-3C2) in unwounded cells, but no migration was observed, only floating cells were observed on the gap. Nonetheless, change in cellular morphology was observed, and limited tumor metastases were prevented within the 38-hour analysis period [28].
Proteases belonging to the caspase family are crucial for both the start and completion of apoptosis but also for preserving homeostasis by controlling inflammation and cell death [29]. These caspases are separated into two groups: executioner caspases (caspase-3, -6, and -7) that carry out the death phase of apoptosis and the initiator caspases (caspase-2, -8, -9, and -10), which are the initial group to be activated in response to a signal [18]. As part of normal homeostasis, apoptotic cell death aids in the removal of damaged or unnecessary cells without harming the surrounding tissues or triggering an immunological reaction. Unsurprisingly, the pathophysiology of many human diseases, including cancer, is caused by improper caspase and apoptosis regulation [30]. Because apoptotic cell death is a natural process and has fewer undesirable side effects, many researchers opt to study the compounds and medications that promote apoptotic cellular death. The results obtained from the Caspase-Glo assay showed a significant upregulation of caspases 3 and 7 (Fig. 10) of MIA PaCa-2 cells treated with methylglyoxal and the IC50 methanolic extract in comparison to untreated cells and DMSO treated cells. Methylglyoxal known for inducing apoptotic cellular death had reading of 19088 RLU, while the IC50-methanolic extract had reading of 9132 RLU (Fig. 10). The potential activation and upregulation of apoptosis is indicated by an increase in RLUs. These results suggest that the plant methanolic extract may have triggered apoptosis, although more research is required to confirm this theory.
Hoechst staining was conducted as further analysis of the kind of cellular death and apoptosis induced by the plant extract. Hoechst staining, in contrast to other assays, attaches to the minor groove of double-stranded DNA (particularly A-T bonds), facilitating the exact detection of nuclear alterations such as condensation and fragmentation, which are typical indicators of apoptotic cell death [31]. Hoechst staining aids in the identification of detrimental mitotic catastrophe frequently triggered by chemotherapy or radiation [31]. It is observable from the obtained Hoechst staining fluorescence microscopic images that the untreated cells (Fig. 13C) emit more fluorescence and maintain their cellular morphology. This was anticipated as their fluorescence dramatically rises when DNA binds, guaranteeing a high signal-to-noise ratio. From the images (Fig. 13D), there are observable cellular changes that IC50-methanolic LP-extract induced apoptotic cellular death as cells underwent chromatin condensation and nuclear fragmentation (lack of blue fluorescence emission) which are morphological markers of apoptosis. This was also observed with the positive control group cells (1% methylglyoxal) (Fig. 13B) which is known to induce apoptotic cell death. The same morphological changes were observed in the negative control group (0.1% DMSO) (Fig. 13A), which was unexpected as DMSO is a universal solvent known to be non-toxic in low concentrations (<10%). At low doses (<0.067), DMSO has been demonstrated to improve cell viability, but at moderate to high concentrations (>0.5), DMSO can exhibit harmful effects. Even though 0.1% DMSO was employed, DMSO is known to change cell membrane permeability, which would affect cell viability and homeostasis [27]. It is also known that DMSO can interact with chemotherapeutic agents to generate adducts that change the toxicity and efficacy, DMSO can also affect mitochondrial respiration, which could result in a rise in reactive oxygen species that could harm cells [27]. As a result of erroneous positives, more testing was necessary.
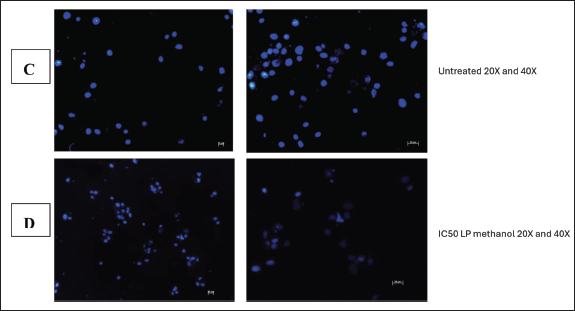 | Figure 13. Image illustration of Hoechst staining of 0.1% DMSO (A) and 1% methylglyoxal (B) treated MIA PaCa-2 cells at 20X and 40X. [Click here to view] |
Mitochondria are widely recognized for their involvement in the synthesis of ATP and the biosynthesis of macromolecules. They are considered the powerhouse of the cell thus the growth, lifespan, and spread of tumors depend on mitochondrial metabolism [32,33]. Moreover, accumulating experimental data supports the notion that mitochondrial dynamics, bioenergetics, and signaling play a vital role in the development of tumors [32]. Research has demonstrated that a variety of cancer cells, namely metastatic tumor cells, cancer stem cells, and therapy-resistant tumor cells, depend on mitochondrial respiration and increase oxidative phosphorylation activity to promote carcinogenesis. Interruption of mitochondrial activity is a trait of programmed cell death in its early phases [33]. Modifications in membrane potential, a crucial component of mitochondrial viability, as well as modifications to the mitochondria’s oxidation-reduction potential are all part of this mitochondrial disturbance [31]. The mitochondria assay is based on the differential detection of biomarkers related to modifications in cellular ATP concentrations and membrane integrity in comparison to vehicle-treated control cells across brief exposure times [31]. ATP assay data analysis was conducted using GraphPad Prism 8.4.3 (686) to generate one-way ANOVA using Dunnett’s comparison test for analysis of ATP Activity of MIA PaCa-2 cells treated with LP-methanolic IC50 crude extract with untreated cells as the control group. From Figure 11, it can be deduced that there was significantly lower ATP detected on MIA PaCa-2 cells treated with LP-methanolic IC50 and positive control group (methylglyoxal) as compared to the untreated cells. This was anticipated as previous procedures (Hoechst staining) showed possible chromosomal damage and chromatin condensation indicating possible mitochondrial damage.
Cells can undergo different types of cellular deaths including apoptosis, autophagy, necrosis, and pyroptosis. However, apoptosis is usually the preferred method of cellular death because it is signal-regulated, non-destructive, and contributes to the maintenance of homeostasis and the normal development of an organism [33]. Cells undergoing apoptosis will typically display membrane shrinkage, chromatin condensation, creation of apoptotic bodies, cytoplasmic condensation, plasma membrane blebbing, and disintegration of the cytoskeleton, with nuclear alteration (nuclear pyknosis) being the most prominent change [17]. Apoptosis is a type of programmed cell death that results in the breakdown of nuclear DNA into several of about 200 bp oligonucleosomal size pieces of which nuclear DNA extraction, and gel electrophoresis characterization of such oligonucleosomal ladders are key methods for detecting apoptosis in cultured cells [34]. In many cells, the biochemical signature of apoptosis is the development of discrete DNA 45 fragments with oligonucleosomal sizes ranging from 180 to 200 by lengths [34]. Large DNA fragments and even single-strand cleavage events may occur after cell death, according to recent discoveries [6,35]. It was observed with the agarose gel (Fig. 14) that the different control groups did not migrate much, which is an indication of large DNA fragments. Quantitative (real-time) PCR (qPCR) is defined as a real-time method of monitoring the development of a PCR reaction as small quantity of PCR product (cDNA, DNA, or RNA) can be measured at the same time [35]. qPCR is a sophisticated procedure for amplification of DNA sequences exponentially which works by detecting the rise of fluorescence emitted by a reporter molecule as the reaction progresses, process is monitored in “real-time” rather than observation of bands on a gel at the end of the reaction [36]. The fluorescent dye or fluorescent-labeled oligos are utilized to determine the quantity of nucleic acid contained in a particular test sample [36]. Upon binding of the dye or probe to the target template, it liberates fluorochrome, which creates fluorescence that can be detected by the detector. A signal is captured by the detector as a positive template amplification.
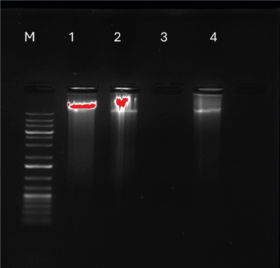 | Figure 14. Agarose gel illustration of DNA fragmentation detection of MIA PaCa-2 cells with different control groups. Lane N: standard molecular size marker (1 Kb). Lane 1: untreated cells, Lane 2: DMSO-treated cells, Lane 3: 1% methylglyoxal treated cells, and Lane 4: LP-methanolic IC50 crude extract. [Click here to view] |
From the graphs (Fig. 15), MIA PaCa-2 cells treated with methanolic IC50 concentration showed significantly high expression levels of genes TP53, RB1, and CHEK1. It can be implied that the methanolic IC50 extract works by signaling DDR pathways, cell cycle regulation, and apoptosis due to upregulation of TP53. TP53, RB1, and CHEK1 are critical tumor suppressor genes that are involved in DNA damage response (DDR), cell cycle regulation, and maintaining genomic stability [28]. Bcl-2 gene, which is known to inhibit p53-mediated death signaling, is downregulated as observed in the graph (Fig. 15). Because TNF (tumor necrosis factor) is upregulated and bcl-2 is downregulated, it is possible that the plant methanolic extract triggers the extrinsic apoptosis pathway by upregulating the FAS gene, which is known to activate caspase 8, which in turn activates caspase 3, 6, and 7 (Fig. 10), resulting in a tumor suppressive mechanism by apoptosis [37]
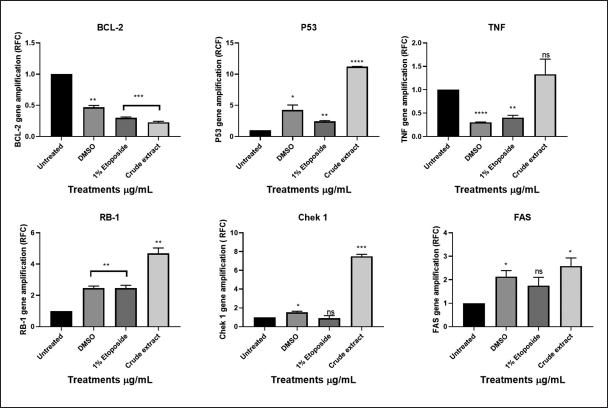 | Figure 15. Graphical illustration of the apoptosis and cell cycle genes (BCL2, TP53, TNF, RB1, CHEK1, and FAS) amplifications deduced with GraphPad Prism 8.4.3 (686). [Click here to view] |
Studies have shown CHEK1 expression levels to be controlled by RB1 and p53 at the transcriptional stage [38]. Common cancer treatments like radiation and chemotherapy do result in several DNA lesions, which trigger the DNA damage response mechanism (DDR) [38]. CHEK1 (critical effector in DDR-DNA damage response) is an essential gene for cellular response to genotoxic chemicals and serves as a biomarker for radiation resistance and is an important part of all cell cycle checkpoints in response to DNA damage by mediating cell cycle arrest [39]. The G2 checkpoint is triggered in response to DNA damage through two different methods [39]. The first is that CHEK1 phosphorylates Cdc25c, which causes it to degrade, consequently, the Cdc2/cyclin B complex is not activated [39]. Second, phosphorylation of the WEE1 kinase results in the inactivation of the Cdc2/cyclin B complex, for p53-mutant cells, this G2 checkpoint blocking is particularly crucial [37]. A promising therapeutic target for the treatment of cancer is the cell-cycle checkpoint kinase WEE1 (Fig. 16) which regulates the G2/M checkpoint and is activated in response to DNA damage caused by CHEK1 kinase [37].
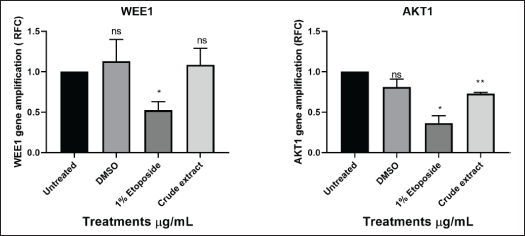 | Figure 16. Graphical illustration of the apoptosis and cell cycle genes (WEE1 and AKT1) amplifications deduced with GraphPad Prism 8.4.3 (686). [Click here to view] |
TP53 is the gene that codes for the p53 tumor suppressor protein while RB1 is renowned as the first identified tumor suppressor gene [40]. Upregulation of these genes (Fig. 15) could possibly indicate activation of apoptosis. FAS (death receptor) is a gene involved in apoptosis generally found on the cell surface and part of the TNF (tumor necrosis factor) superfamily. TNF genes control inflammation; thus, elevated levels of TNF could indicate more inflammation and low levels of TNF could indicate less inflammation [37]. The one-way ANOVA test showed insignificant levels of TNF in comparison to untreated cells [37]. AKT1 genes are crucial for controlling a variety of cellular functions, including growth, survival, and metabolism, through the PI3K/Akt signaling pathway, which is frequently dysregulated by cancer cells, particularly pancreatic cancer [40]. AKT1 is commonly overexpressed or highly active in pancreatic cancer cells, which aids in the development of cancer by boosting proliferation, aiding metastasis, boosting cell survival, and raising drug resistance, among other effects [40]. Significantly higher levels of AKT1 (Fig. 16) were found in the experiment, but other studies suggested that the plant extract may have activated apoptosis and metastasis [41]. AKT1 plays a complex role and situation-specific involvement in cell invasion and migration [40]. Several cancers, such as ovarian, lung, and pancreatic tumors, have been found to have aberrant overexpression or activation of AKT, which is linked to enhanced cancer cell survival and proliferation [17]. As a result, AKT targeting may offer a crucial strategy for cancer treatment and prevention [41]. Furthermore, it has been demonstrated that AKT1 inhibits the epithelial-to-mesenchymal transition (EMT), a process linked to enhanced invasiveness in some malignancies, including breast cancer [26]. On the other hand, AKT1 might encourage invasion and migration in other circumstances [40]. The MIA PaCa-2 cell line was discovered to be an incredibly aggressive cancer cell line in this study because it grew quickly and was able to successfully migrate into the artificial wound in just 24 hours (Fig. 9-C1). Thus, given what is known about AKT1 from the literature, the MIA PaCa-2 cell line’s overexpression of this gene is not unexpected. This gene may possibly be exploited to detect cancer metastases or serve as the target gene for the development of therapeutic drugs.
CONCLUSION
Pancreatic cancer is one of the most aggressive forms of cancer with a high mortality rate. It is ranked 12th most common and 6th deadliest type of cancer. When it is lately detected, it can be lethal with no reliable therapeutic agent insight. In this study, we explored the crude methanolic extract of L. petersonii as a potential anticancer and antimetastasis agent. Because certain protocols lacked standardized procedures, it was challenging to compare them with earlier research due to optimization and modifications. To validate and rule out unsubstantial findings, various techniques and tests were conducted. Scientific results were obtained after various laboratory methods and techniques were established. Certain protocols could not be repeatedly performed due to time, resource limitations, and access restrictions with sophisticated equipment. From the results obtained, an active compound was isolated which we will further characterize and explore its anticancer activity. We further showed that L. petersonii crude extract is cancer-type-dependent and, in some cases, concentration-dependent. In terms of molecular mechanism, the results suggest that apoptosis might be activated. However, further studies would need to be carried out to confirm the test in in vivo studies.
ACKNOWLEDGMENTS
The authors would like to thank Dr Michael Hermann Kamdem Kengne for helping in understanding and compiling the chemistry part of the project, without his support it would not have been possible.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
FINANCIAL SUPPORT
This research was funded by the National Research Foundation, grant number PMDS22072042467.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY STATEMENT
All the data is available with the authors and shall be provided upon request.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
PUBLISHER’S NOTE
All claims expressed in this article are solely those of the authors and do not necessarily represent those of the publisher, the editors and the reviewers. This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
USE OF ARTIFICIAL INTELLIGENCE (AI)-ASSISTED TECHNOLOGY
The authors declare that they have not used artificial intelligence (AI)-tools for writing and editing of the manuscript, and no images were manipulated using AI.
REFERENCES
1. Seraphin TP, Joko-Fru WY, Kamaté B, Chokunonga E, Wabinga H, Somdyala NI, et al. Rising prostate cancer incidence in sub-Saharan Africa: a trend analysis of data from the African cancer registry network. Cancer Epidemiol Biomarkers Prev. 2021;30(1):158–65. CrossRef
2. Priya S, Satheeshkumar P. Natural products from plants: recent developments in phytochemicals, phytopharmaceuticals, and plant-based neutraceuticals as anticancer agents. Funct Preserv Prop Phytochem. 2020;2020:145–63. CrossRef
3. Clinton SK, Giovannucci EL, Hursting SD. The world cancer research fund/American institute for cancer research third expert report on diet, nutrition, physical activity, and cancer: impact and future directions. J Nutr. 2020;150(4):663–71. CrossRef
4. Banerjee S, Kaviani A. Worldwide prostate cancer epidemiology: differences between regions, races, and awareness programs. Int J Clin Exp Med Sci. 2016;2:1–6. CrossRef
5. Nhleko M, Edoka I, Musenge E. Pancreatic cancer mortality in South Africa: a case-control study. S Afr Med J. 2024;114(1):27–32. CrossRef
6. Muhammad N, Usmani D, Tarique M, Naz H, Ashraf M, Raliya R, et al. The role of natural products and their multitargeted approach to treat solid cancer. Cells. 2022;11(14):2209. CrossRef
7. World Health Organization. Global cancer burden growing, amidst mounting need for services. Geneva: World Health Organization; 2024.
8. Mothibe ME, Sibanda M. African traditional medicine: South African perspective. Trad Complem Med. 2019;2019:1–27. CrossRef
9. Adico MD, Bayala B, Bunay J, Baron S, Simpore J, Lobaccaro J-MA. Contribution of sub-Saharan African medicinal plants to cancer research: scientific basis 2013–2023. Pharmacol Res. 2024:107138. CrossRef
10. Borkar, S. 2023. Rf Value: calculation, significance and affecting factors. Available from: https://www.chemistrywithdrsantosh.com/2023/05/Rf-value-calculation-significance-and-affecting-factors.html
11. Sasidharan S, Chen Y, Saravanan D, Sundram K, Latha LY. Extraction, isolation and characterization of bioactive compounds from plants’ extracts. Afr J Tradit Complement Altern Med. 2011;8(1):1–10. CrossRef
12. Bajpai VK, Majumder R, Park JG. Isolation and purification of plant secondary metabolites using column-chromatographic technique. Bangladesh J Pharmacol. 2016;11(4):844–8. CrossRef
13. Mohd Rehan S, Ansari FA, Singh O. Isolation, identification, antibacterial activity and docking of fatty acid and fatty alcohol from rumex dentatus leaf extract. Int J Pharm Sci Rev Res. 2020;64(1):7–11. CrossRef
14. Saifullah M, McCullum R, McCluskey A, Vuong Q. Comparison of conventional extraction technique with ultrasound assisted extraction on recovery of phenolic compounds from lemon scented tea tree (Leptospermum petersonii) leaves. Heliyon. 2020;6(4):e03666. CrossRef
15. Pascual M, Carretero M, Slowing K, Villar A. Simplified screening by TLC of plant drugs. Pharm Biol. 2002;40(2):139–43. CrossRef
16. Kumar A, P N, Kumar M, Jose A, Tomer V, Oz E, et al. Major phytochemicals: recent advances in health benefits and extraction method. Molecules. 2023;28(2):887. CrossRef
17. Pina-Sanchez P, Chavez-Gonzalez A, Ruiz-Tachiquin M, Vadillo E, Monroy-Garcia A, Montesinos JJ, et al. Cancer biology, epidemiology, and treatment in the 21st century: current status and future challenges from a biomedical perspective. Cancer Control. 2021;28:10732748211038735. CrossRef
18. Arandjelovic S, Ravichandran KS. Phagocytosis of apoptotic cells in homeostasis. Nat Immunol. 2015;16(9):907–17. CrossRef
19. Abdel-Tawab M. Considerations to be taken when carrying out medicinal plant research—what we learn from an insight into the IC50 values, bioavailability and clinical efficacy of exemplary anti-inflammatory herbal components. Pharmaceuticals. 2021;14(5):437. CrossRef
20. Canga I, Vita P, Oliveira AI, Castro MÁ, Pinho C. In vitro cytotoxic activity of african plants: a review. Molecules. 2022;27(15):4989. CrossRef
21. Somaida A, Tariq I, Ambreen G, Abdelsalam AM, Ayoub AM, Wojcik M, et al. Potent cytotoxicity of four cameroonian plant extracts on different cancer cell lines. Pharmaceuticals. 2020;13(11):357. CrossRef
22. Veß A, Hollemann T. Methylglyoxal induces nuclear accumulation of p53 and γH2AX in normal and cancer cells. bioRxiv. 2020:2020.03. 19.998773. CrossRef
23. Tung MHT, ?uc HV, Huong TT, Duong NT, Tai BH, Kim YH, et al. Cytotoxic compounds from Brucea mollis. Sci Pharm. 2012;81(3):819. CrossRef
24. Grada A, Otero-Vinas M, Prieto-Castrillo F, Obagi Z, Falanga V. Research techniques made simple: analysis of collective cell migration using the wound healing assay. J Investig Dermatol. 2017;137(2):e11-e6. CrossRef
25. Iqbal J, Abbasi BA, Mahmood T, Kanwal S, Ali B, Shah SA, et al. Plant -derived anticancer agents: a green anticancer approach. Asian Pac J Trop Biomed. 2017;7(12):1129–50. CrossRef
26. Pijuan J, Barceló C, Moreno DF, Maiques O, Sisó P, Marti RM, et al. In vitro cell migration, invasion, and adhesion assays: from cell imaging to data analysis. Front Cell Dev Biol. 2019;7:107. CrossRef
27. Hoang BX, Han B, Fang WH, D TRAN H, Hoang C, Shaw DG, et al. The rationality of implementation of dimethyl sulfoxide as differentiation-inducing agent in cancer therapy. Cancer Diagn Prog. 2023;3(1):1. CrossRef
28. Guo Y, Zhang Y, Yang X, Lu P, Yan X, Xiao F, et al. Effects of methylglyoxal and glyoxalase I inhibition on breast cancer cells proliferation, invasion, and apoptosis through modulation of MAPKs, MMP9, and Bcl-2. Cancer Biol Ther. 2016;17(2):169–80. CrossRef
29. McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harbor Perspect Biol. 2013;5(4):a008656. CrossRef
30. Boice A, Bouchier-Hayes L. Targeting apoptotic caspases in cancer. Biochim Biophys Acta. 2020;1867(6):118688. CrossRef
31. Kiraly G, Simonyi AS, Turani M, Juhasz I, Nagy G, Banfalvi G. Micronucleus formation during chromatin condensation and under apoptotic conditions. Apoptosis. 2017;22:207–19. CrossRef
32. Porporato PE, Filigheddu N, Pedro JMB-S, Kroemer G, Galluzzi L. Mitochondrial metabolism and cancer. Cell Res. 2018;28(3):265–80. CrossRef
33. Kabakov AE, Gabai VL. Cell death and survival assays. Chaperones. 2018;2018:107–27. CrossRef
34. Saadat YR, Saeidi N, Vahed SZ, Barzegari A, Barar J. An update to DNA ladder assay for apoptosis detection. BioImpacts. 2015;5(1):25. CrossRef
35. Chauhan T. Real-Time PCR: principle, procedure, advantages, limitations and applications. Genet Educ PCR Technol. 2019;22:2023.
36. Aryal S. Real time PCR-principle, process, markers, advantages, uses; 2021. [cited 2021 March 4]. Available from: https://microbenotes com/real-time-pcr-principle-process-markers-advantages-applications/
37. Van Loo G, Bertrand MJ. Death by TNF: a road to inflammation. Nat Rev Immunol. 2023;23(5):289–303. CrossRef
38. Bargiela-Iparraguirre J, Prado-Marchal L, Fernandez-Fuente M, Gutierrez-González A, Moreno-Rubio J, Munoz-Fernandez M, et al. CHK1 expression in Gastric Cancer is modulated by p53 and RB1/E2F1: implications in chemo/radiotherapy response. Sci Rep. 2016;6(1):21519. CrossRef
39. Wang Z, Li W, Li F, Xiao R. An update of predictive biomarkers related to WEE1 inhibition in cancer therapy. J Cancer Res Clin Oncol. 2024;150(1):13. CrossRef
40. Li C-W, Xia W, Lim S-O, Hsu JL, Huo L, Wu Y, et al. AKT1 inhibits epithelial-to-mesenchymal transition in breast cancer through phosphorylation-dependent Twist1 degradation. Cancer Res. 2016;76(6):1451–62. CrossRef
41. Zhang J, Gan Y, Li H, Yin J, He X, Lin L, et al. Inhibition of the CDK2 and Cyclin A complex leads to autophagic degradation of CDK2 in cancer cells. Nat Commun. 2022;13(1):2835. CrossRef