
INTRODUCTION
Breast cancer, the second most common cancer disease globally, accounts for a substantial number of annual deaths among women [1]. Breast cancer development is influenced by both genetic and epigenetic changes, which can disrupt signaling pathways and contribute to tumor growth. In addition, alterations in the tumor micro-environment can promote invasion and metastasis processes [2]. Breast cancer cells are characterized into distinct subtypes based on the expression of human epidermal growth factor receptor 2 (HER2), estrogen receptor (ER), and progesterone receptor (PR). These molecular subtypes are clinically divided into major forms that include Luminal A (ER+ and/or PR+ or HER2-), Luminal B (ER+ and/or PR+ or PR- and/or HER2+/-), HER2-enriched, and basal/triple-negative breast cancer (TNBC) [2]. In breast cancer, up to 70% of the cells show positive immunohistochemical detection for ER and/or PR receptors. Other subtypes, such as HER2 comprise approximately 12%–20%, while TNBC accounts for 15%–20% of invasive breast cancer cases [3]. Breast cancer treatment mostly involves surgery, radiation therapy, immunotherapy, and chemotherapy [4]. Chemotherapy, a standard treatment for cancer, specifically targets the rapid proliferation of cancer cells. However, it also affects normal cells that undergo rapid division, leading to undesired side effects and potential damage to some organs [5]. In response to these challenges, extensive research is being conducted to identify and develop new anticancer agents derived from natural products. These efforts aim to discover compounds that may offer more targeted and less toxic therapeutic options for cancer treatment.
Curcumin is a well-known example of a natural compound with significant clinical promise due to its anti-inflammatory, antioxidant, and anticancer properties [6]. Despite its broad activity spectrum and great safety profile, curcumin faces challenges in its development as a medication due to its poor oral bioavailability [7], which is characterized by poor absorption, limited water solubility, and rapid metabolism. In addition, the instability of curcumin in alkaline conditions and systemic elimination further limit its application as an effective therapeutic agent [8,9]. Consequently, there is ongoing interest in developing curcumin analogs that preserve its biological efficacy while improving the oral bioavailability properties. Notably, the presence of an active methylene group and α,β-diketone moiety contribute to curcumin instability and poor absorption under physiological conditions [10].
The development of new anticancer agents from natural products also involves bioavailability improvement and pharmacological activity enhancement through chemical modifications. The synthesis of curcumin analogs aims to obtain more effective treatments for various diseases. Heterocyclic compounds play a crucial role in medicinal chemistry, as some biologically active molecules, including vitamins, hormones, enzymes, antibiotics, nucleic acids [Deoxyribonucleic acid (DNA) and Ribonucleic acid], hemoglobin, and chlorophyll, contain heterocyclic rings. These organic compounds typically incorporate at least one heteroatom, commonly oxygen, nitrogen, or sulfur on their cyclic structures [11]. The ring structure not only enhances the stability of the molecule but also improves solubility, facilitating oral absorption and bioavailability [12]. The 1,3-dicarbonyl site is particularly attractive for integrating heterocyclic groups to create novel and stable derivatives, making the unique structure of curcumin as an excellent candidate for this approach.
Various studies have been on the synthesis of curcumin analogs that involve modifications to the structure of curcumin, including the introduction of mono-carbonyl groups and substitutions on the aromatic ring. This modification has been proven to increase the biological activity. Al-Hujaily et al. [13] synthesized 5-bis(4-hydroxy-3-methoxybenzylidene)-N-methyl-4-piperidone (PAC) by replacing the methylene group and the two carbonyl groups in curcumin with N-methyl-4-piperidone, which increased both stability and hydrophilicity. At a concentration of 40 μM, PAC induced cell death in 35% of MCF-7 cells and 70% of T47D cells, whereas curcumin triggered apoptosis in less than 20% of both cells. In addition, Adams et al. [14] prepared and screened a series of curcumin analogs, among which 3,5-bis((E)-2-fluorobenzylidene)piperidin-4-one exhibited significant cytotoxicity and antiproliferative properties against MDA-MB-231 and PC3 cancer cell lines, demonstrating potency greater than curcumin. Based on those previous studies, we developed novel anticancer agents of mono-carbonyl curcumin analogs and evaluated their anticancer effects against TNBC 4T1, HER2 enriched group, Luminal A T47D, and MCF-7 cells. In addition, in silico studies the three most promising compounds were conducted using molecular docking to reveal their interactions with protein receptors identified through bioinformatics, along with predictions of their pharmacokinetic profiles.
MATERIALS AND METHODS
Materials
The reagents, including 3-bromo-4-methoxybenzaldehyde, N-benzyl-4-piperidone, 4-piperidone, and N-methyl-4-piperidone, were purchased from Macklin in pro-analysis specification. Ethanol and sodium hydroxide were purchased from Merck in pro analytical grade. The materials used in the cytotoxicity activity include 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), Modified Eagle Medium, fetal bovine serum, trypsin-EDTA 0.25%, phosphate buffer saline, sodium dodecyl sulfate (SDS), and HCl 0.01 M. The cell cultures used are T47D, HER2, 4T1, and MCF-7 for cancer cell culture, as well as Vero for normal cell culture, with doxorubicin as the positive control.
Instrumentation
The equipment used in this research includes ELISA reader (Bio-Rad, Benchmark), melting point apparatus (Electrothermal IA9100), TLC scanner (CAMAG TLC Scanner), FTIR-ATR spectrophotometer (Shimadzu Prestige-21), 1H-NMR (500 MHz), and 13C-NMR (125 MHz) (JEOL JNMECA 500).
General synthesis of compounds A, B, and C
The synthesis process was conducted using a modified method from the previous work [15]. This involved dissolving 3-bromo-4-methoxybenzaldehyde (2 mmol) in 10 ml of ethanol and mixing it with 1 mmol of piperidone derivatives (N-benzyl-4-piperidone, 4-piperidone, and N -methyl-4-piperidone). The solution was stirred at room temperature for 30 minutes, followed by a drop-wise addition of NaOH solution (1 ml of 20% NaOH for compound A; 2 ml of 30% NaOH for compound B; and 1 ml of 30% NaOH for compound C). The solution was then subjected to ultrasonic irradiation for 1, 7, and 2 hours to produce a yellow precipitate of compounds A, B, and C, respectively. The yellow solid of compound A was recrystallized with ethanol and then dried under vacuum. Table 1 lists the characterization and elucidation data of compounds A–C.
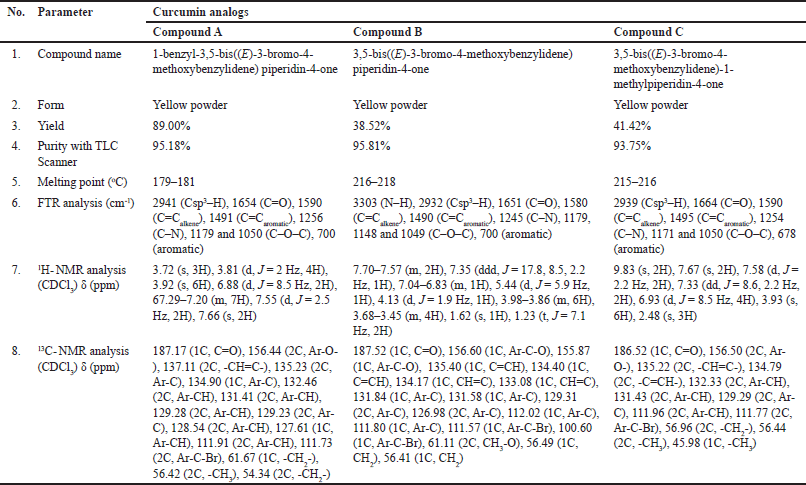 | Table 1. Characterization and elucidation of the structure of curcumin analog compounds. [Click here to view] |
Cytotoxic activity
The cytotoxic activities were measured by the MTT assay using a modified method from the previous reports [3,16]. Briefly, cells were harvested and plated in a well plate, followed by incubation for 24 hours before the treatment. Various concentrations of compounds A, B, and C (ranging from 12.5 to 200 µg/ml) were diluted in the culture medium and applied for 24 hours treatments in MCF-7, T47D, 4T1, and HER2 cells. After treatment, the medium was replaced with 100 µl of MTT and incubated for 4 hours to allow for purple formazan crystal formation. When formazan had formed, 100 µl of 10% SDS-HCl solution was added as a stopper and the well plate was stored overnight. The absorbance was recorded with an ELISA reader, while the IC50 and selectivity index values of the synthesized compounds were calculated accordingly.
Pharmacokinetic profile prediction (ADMET)
Pharmacokinetic profile prediction was performed through the swissADME (http://www.swissadme.ch), and ADMETlab 2.0 (http://admet.scbdd.com/) websites by entering the Simplified Molecular Input Line Entry System (SMILES) codes of curcumin and synthesized curcumin analogs, downloaded from the PubChem website (www.pubchem.ncbi.nlm.nih.gov).
Collection and target establishment of curcumin analog compounds
The optimized curcumin analogs were opened using ChemDraw to obtain SMILES data, then they were uploaded to the Swiss Target Prediction database (http://www.swisstargetprediction.ch/). The predicted target gene data were downloaded in CSV format, filtered, and integrated using Microsoft Excel 365 software [17,18]. In addition, disease-related targets were retrieved by searching the keyword “breast cancer” from four public databases: National Center for Biotechnology Information GeneCards database (https://www.genecards.org/), Online Mendelian Inheritance in Man (OMIM, https://omim.org/), Therapeutic Target Database (TTD, http://db.idrblab.net/ttd/), and DisGeNET (https://www.disgenet.org/). Both the curcumin analog compound gene and disease target gene were integrated with VENNY 2.1 (https://bioinfogp.cnb.csic.es/tools/venny/index.html) [19,20].
Protein–protein interaction (PPI) data screening and analysis
Each gene of curcumin analog compounds and integrated disease targets built PPI relationships by uploading to the STRING data site (https://string-db.org/) with the Homo sapiens setting, a confidence level of 0.4, and other parameters selected as default. PPI relationships with node1, node2, and the combined score from the export file were imported into Cytoscape 3.10.2 software to form an interaction network. The results of the top target analysis were performed using the CytoHubba plugin. This step is used to build and visualize networks, especially in the field of network pharmacology analysis [19,20,18].
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGGs) enrichment analysis
Gene Ontology and KEGG pathway analysis were performed to further study the functions of identified target genes, including biological processes (BPs), cellular components (CCs), gene functionality, CC, and molecular functions (MFs). The results of STRING data bioinformatics analysis were uploaded to ShinyGO 0.82 data (http://bioinformatics.sdstate.edu/go/) with the “Human” setting to obtain combined targets. The main target proteins were validated by molecular docking analysis.
Molecular docking
Proteins obtained from the bioinformatic approach were traced through the website www.rcsb.org to obtain the PDB ID. Protein structures and standard ligands were prepared using Autodock tools. Curcumin and its analogs were modeled and optimized using GaussView 5.0 software with density functional theory, B3LYP hybrid functional, and 3-21G(d,p) basis set. Molecular tethering simulations were performed with Autodock Vina. Root-mean-square deviation (RMSD) values were obtained by performing re-docking studies of the native protein and each ligand prepared in a 30 Å cubic grid box (PDB ID 2G01 and 4EJN) with a spacing of 0.375 Å for 100 runs of the generic Lamarck algorithm using AutoDock4.2 software. The re-docking process was declared valid if the RMSD value was lower than 2.00 Å. The molecular docking studies were performed using the same parameters as the re-docking investigation. Parameters reviewed in molecular tethering were affinity energy (increasingly negative values indicate higher stability), RMSD value, and interaction between ligand and receptor protein. The interaction of the most stable conformation was visualized using BIOVIA Discovery Studio 2019 Client software [21].
RESULTS AND DISCUSSION
Synthesis of curcumin analogs
The chemical modification of curcumin involves the replacement of α,β-diketone with mono-carbonyl. Three novel mono-carbonyl analogs of curcumin, i.e., N-benzyl-4-piperidone (A), 4-piperidone (B), and N-methyl-4-piperdone (C), were designed by displacing α,β-diketone moiety with a single carbonyl group. The synthesis and structures of compounds A–C are shown in Figure 1. The target compounds were synthesized through the direct aldol condensation of substituted benzaldehyde with the ketones in alkaline media. The synthesized compounds were characterized using FTIR, 1H-NMR, and 13C-NMR spectroscopies, confirming the correct structures of compounds A–C. The FTIR spectra of compounds A, B, and C, shown in Figure 2, exhibit absorption signals at 3,303, 2,941, 1,654, 1,590, 1,491, 1,256, and 1,179 cm?¹, corresponding to the stretching vibrations of N–H, Csp³–H, C=O, C=Calkene, C=Caromatic, C–N, and C–O–C functional groups, respectively. The mono-carbonyl curcumin analogs A–C are distinguished by peaks in the range of 2,941–2,932 cm?¹, which correspond to C–H stretching vibrations. In addition, the C–N stretching is indicated by peaks in the range of 1,256–1,245 cm?¹. Furthermore, the structures of curcumin analogs were confirmed by analyzing the hydrogen splitting patterns in the 1H-NMR spectra and the number of carbon signals in the 13C-NMR spectra. The spectra show a C=O signal at δ 186–187 ppm and CH aromatic signals at δ 131–132 ppm on their 13C-NMR spectra. In addition, the C–Br signal appears at δ 111–112 ppm, and the C–O signal at δ 156 ppm is more de-shielded than the C–Br signals due to the electron-withdrawing effect of the oxygen atom on their 13C-NMR spectra. These characterizations provide strong evidence for the successful synthesis of the mono-carbonyl curcumin analogs A–C.
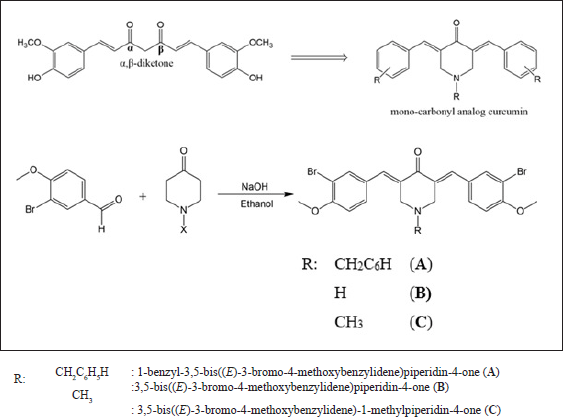 | Figure 1. The synthesis scheme of curcumin analogs. [Click here to view] |
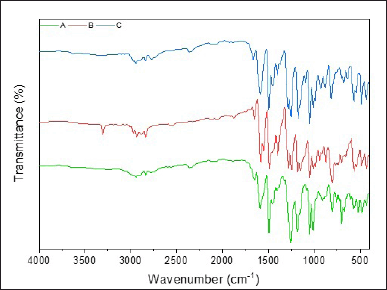 | Figure 2. The FTIR spectra of curcumin analogs A, B, and C. [Click here to view] |
Pharmacokinetic
A drug candidate must reach its inhibitory target at sufficient concentrations, and it must remain at the active site for a period to generate the expected biological effects. Undesirable pharmacokinetic properties or intolerable toxicity levels are leading causes of failure in the development of new drug candidates during clinical trials. Therefore, the development of new drug candidates must include the ADMET evaluations to ensure high efficacy and safety as therapeutic agents.
To evaluate the absorption properties of compounds A–C, we predict Caco-2 permeability and human intestinal absorption (HIA), as presented in Table 2. The Caco-2 cell assay is a key method for evaluating passive and active transports and the absorption of orally administered drugs. The predicted values of Caco-2 permeability range from −4.732 to −6.63 cm/second, suggesting low to moderate permeability [22]. Compounds B and C show low Caco-2 permeability, similar to curcumin, indicating that these compounds may have low absorption. In contrast, compound A exhibits higher Caco-2 permeability than curcumin, suggesting that it may have better absorption. HIA is a crucial parameter for the efficacy of drug compounds because it is closely related to bioavailability, as it is reported that 64% of a compound’s bioavailability is significantly influenced by HIA [23]. High HIA values, expressed as %absorption, indicate that the drug compound is well absorbed in the intestine, while compounds with an absorption rate of less than 30% are considered poorly absorbed [22]. It is shown that compounds A–C exhibit higher absorption than curcumin, suggesting that the proposed mono-carbonyl curcumin analogs will provide improved absorption compared to curcumin.
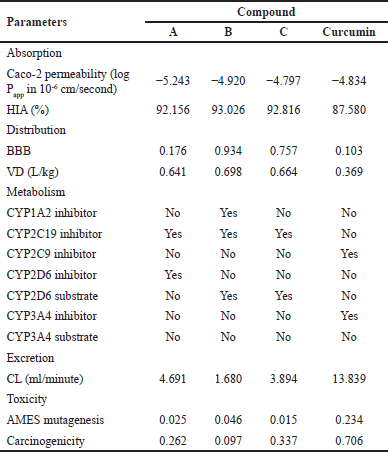 | Table 2. Prediction of pharmacokinetics for curcumin analogs A–C and curcumin. [Click here to view] |
Blood–brain barrier (BBB) permeability and volume of distribution (VD) are key parameters in studying the distribution of drug candidates. The ability of a drug compound to penetrate the brain is crucial for mitigating potential side effects. The BBB plays a significant role in the absorption of drug compounds into the brain. Molecules with a logBB > −1 are classified as BBB+, indicating they can penetrate the BBB, while those with logBB ≤ −1 are classified as BBB-, indicating their low ability to penetrate BBB. Based on Table 2, compounds B and C can easily pass through the BBB, whereas compound A and curcumin could not. Consequently, compounds B and C can be distributed to the central nervous system, while it is not possible for compound A. For drug candidates targeting breast cancer, it is generally preferable for them not to cross the BBB, as the target is located in cancer tissue. Compound A has a relatively moderate BBB value, suggesting that a small amount may still pass through the barrier, raising the possibility that it could reach the target cancer tissue.
The volume of distribution is a measure that indicates how far a drug spreads in the body after administration. VD values provide insights into how well the drug binds to plasma protein, its distribution in body fluids, and its absorption by tissues. A compound is considered to have an appropriate VD if its value is within the range of 0.04–20 L/kg [24]. Compounds A–C exhibit a larger distribution volume compared to curcumin (Table 2). Generally, drug compounds with high lipophilicity are more likely to bind to body tissues rather than remain in blood plasma, resulting in a lower concentration in plasma and a larger distribution volume. Compounds A–C are predicted to be distributed less in the blood but more in tissues than curcumin. This suggests that the prepared compounds have a superior volume of distribution compared to curcumin, leading to higher tissue distribution.
Drug metabolism parameters in the body measure how the enzymatic system breaks down drugs and determines the duration and intensity of their action. A crucial aspect is to avoid the inhibition of cytochrome P450 (CYP450) enzymes. Approximately 80% of this metabolic activity is attributed to five key isozymes, named 1A2, 3A4, 2C9, 2C19, and 2D6 [25]. Most of these CYP enzymes, responsible for phase I metabolic reactions, are concentrated in the liver, the primary organ for drug metabolism. Phase I drug-metabolizing enzymes, particularly members of the CYP family, are responsible for the oxidation of many drugs [26]. When these enzymes are inhibited, the metabolic clearance of their substrate drugs decreases, leading to higher concentrations of the drugs in the body, which can increase the risk of toxicity by allowing drug levels to exceed their therapeutic window. Predictions regarding drug metabolism indicate that compounds B and C are metabolized by the CYP2D6 enzyme, whereas none of the compounds are predicted to interact with CYP3A enzymes. Compounds A–C inhibit CYP450 enzyme, suggesting they may inhibit phase I metabolism more effectively than curcumin, thereby increasing bioavailability in the body.
Clearance (CL) refers to the body’s ability to eliminate a drug from its system. It is a crucial factor in determining the duration a drug remains in the body (half-life) and the frequency at which doses should be administrated. Based on Table 2, the CL values of compounds A, B, and C are lower than curcumin, indicating that the elimination process for these three curcumin analogs will take longer. Ensuring a compound is non-toxic is a crucial criterion for choosing a compound as a potential therapeutic candidate. The toxicity levels of curcumin analog compounds were evaluated through AMES mutagenesis and carcinogenicity. The output values presented in Table 2 indicate the probability of toxicity, ranging from 0 (low toxicity) to 1 (high toxicity). The toxicity prediction results for compounds A, B, and C have very low potential mutagenic or carcinogenic effects based on data, even lower than curcumin.
The evaluation of new drug candidates is also conducted by analyzing through Lipinski’s “Rule of Five”, which consists of combined parameters capable of identifying potential drug candidates that may present problems with absorption and permeability [27]. According to this rule, poor oral bioavailability is more likely when there are more than 10 hydrogen bond acceptors, 5 hydrogen bond donors, a molecular weight greater than 500, and a calculated LogP greater than 5 [28]. The results of the analysis of the mono-carbonyl curcumin analog compounds based on Lipinski’s “Rule of Five” are presented in Table 3. Only compound B meets Lipinski’s “Rule of Five” while compounds A and C do not follow the rule, suggesting that these curcumin analogs may have poor oral bioavailability. However, despite this drawback, their bioavailability could potentially be improved through further research and the use of advanced drug delivery systems such as nanoparticles, liposomes, polymeric nanocarriers, lipid-based formulations, or other innovative formulations [27]. These strategies are able to enhance absorption and therapeutic efficacy, allowing for more effective use of these compounds in clinical settings.
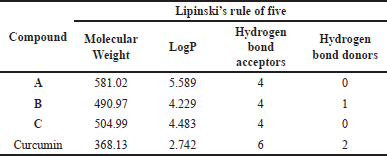 | Table 3. Results of compound analysis based on Lipinski’s rule. [Click here to view] |
Cytotoxicity activity of mono-carbonyl curcumin analog compounds
The cytotoxicity of curcumin analogs was evaluated on breast cancer cell lines T47D, MCF7, HER2, and 4T1 using the MTT assay. The selection of cancer cell lines represents the classification of breast cancer based on immuno-histochemical methods. Luminal A characterized by the expression of ER and PR receptors with negative HER2 status is represented by MCF-7 and T47D cell lines [29]. For the HER2+ subtype, which demonstrates HER2 overexpression without ER/PR expression the HER2 cell line is utilized [30]. In addition, 4T1 represents the characteristics of TNBC, which typically does not express ER, PR, or HER2 [31]. Understanding the differential responses of these cell lines to curcumin analogs can provide insights into their potential therapeutic efficacy and inform treatment strategies for various breast cancer subtypes.
Cytotoxicity is quantified by the IC50 value, defined as the drug concentration required to inhibit cell viability by 50% [32]. Based on the data presented in Table 4, all mono-carbonyl curcumin analogs exhibit high to moderate cytotoxicity against the T47D cells, while showing no cytotoxic effects on the MCF-7 and 4T1 cells. Compound B demonstrates high cytotoxicity toward T47D cell, with an IC50 value of 19.20 µg/ml. In addition, compounds B and C demonstrate moderate cytotoxicity against the HER2 cells. These findings suggest that mono-carbonyl curcumin analogs may selectively target certain breast cancer subtypes, particularly those associated with hormone receptor expression. The high cytotoxicity of compound B underlines its potential as a promising candidate for further development. Understanding the pathways involved in the action of these compounds, especially in relation to the T47D cells, is crucial for elucidating their mechanisms of action and therapeutic potential. Utilizing bioinformatics tools to analyze gene expression profiles and signaling pathways, along with molecular docking studies to predict the interactions between the compounds A–C and breast cancer cell lines. Therefore, valuable insights into how mono-carbonyl curcumin analogs interact with breast cancer cell lines could be provided.
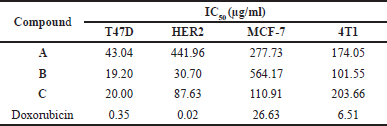 | Table 4. Effect of mono-carbonyl curcumin analogs on viability of breast cancer cell lines. [Click here to view] |
In addition to IC50 values, anticancer activity can be evaluated using the selectivity index (SI), which indicates a drug’s safety and effectiveness. The SI quantifies the extent to which a drug selectively targets cancer cells over normal cells. The SI is calculated from the ratio of the IC50 in normal cells to that of cancer cells. A higher SI indicates that the drug is more specific to cancer cells while minimizing toxicity to normal cells, thereby reflecting a more favorable safety profile [33]. The synthesized compounds A, B, and C demonstrate low selectivity, suggesting that these compounds may exert cytotoxic effects on both cancer and normal cells (Table 5). This low selectivity may lead to adverse side effects that limit the therapeutic potential of these compounds. Consequently, further research is needed to enhance their selectivity. To address these limitations, it is required to employ drug delivery systems that specifically target cancerous tissues.
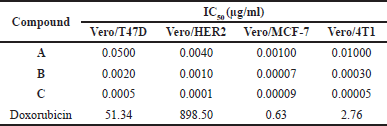 | Table 5. Selectivity index of curcumin analogs. [Click here to view] |
Targets and breast cancer-related targets of curcumin analogs
Drug discovery by utilizing bioinformatic and computational biology plays a pivotal role in the drug discovery progress. Its existence provides information on the molecules’ characterization and chemical behavior of compounds in certain therapeutic processes [34]. The procedure for obtaining bioinformatic information from each curcumin analog against a breast cancer target is presented in Figure 1. The results of the bioinformatic approach from the potential of each curcumin analog are obtained from the Swiss prediction target database. Compounds A, B, and C contain 60, 39, and 27 genes, respectively. Therapeutic targets searched from the GeneCards Human, DisGeNet, OMIM, and TDD databases with the keyword “breast cancer” obtained 13,881 target genes. Each of the curcumin analog was built to obtain potential targets for breast cancer therapy using a Venn diagram (Fig. 3a). Potential target genes that overlap between curcumin analogs and breast cancer were constructed using PPI with the STRING database (Fig. 3b). Further analysis using Cytoscape 3.10.2 was done to obtain the main compound-target network plot focused on pathways in cancer pharmacology. The determination of network pharmacology pathways in cancer is based on GO and KEGG analysis. GO analysis consists of three aspects of enrichment analysis, i.e., BP, MF, and CC. The top 10 of each GO parameter of the compound are shown in Figure 4. KEGG analysis shows the 15 highest network pharmacology pathways, including the pathways in cancer for breast cancer therapy (Fig. 5a). Proteins involved in network pharmacology in cancer pathways in breast cancer therapy are shown in Figure 5b. The set parameters in determining the main protein in the pathways in cancer in breast cancer therapy are the average length of the shortest path, betweenness centrality, closeness centrality, clustering coefficient, edge count, oddness, and inward degree. The main proteins that play a role in therapy (marked in orange) are mitogen-activated protein kinase 8 (MAPK8), AKT1, and AKT1 for curcumin analogs A, B, and C.
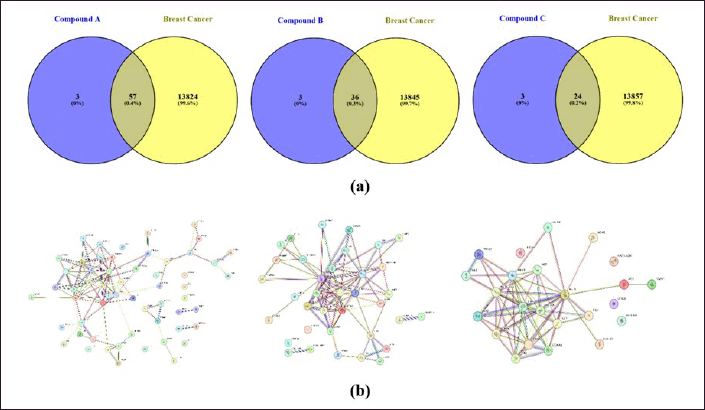 | Figure 3. Bioinformatic information of each curcumin analog compound in breast cancer therapy prediction; (a) Venn diagram; (b) PPI STRING data between breast cancer compound genes. [Click here to view] |
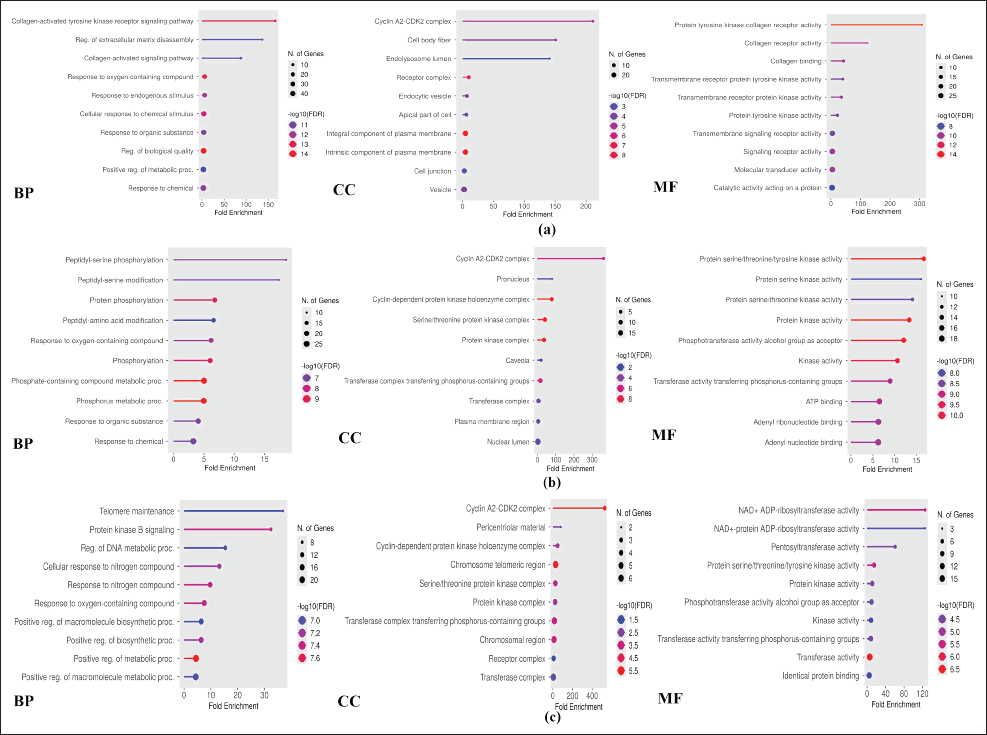 | Figure 4. Top 10 GO analyses of each parameter for compound (a) A, (b) B, and (c) C. [Click here to view] |
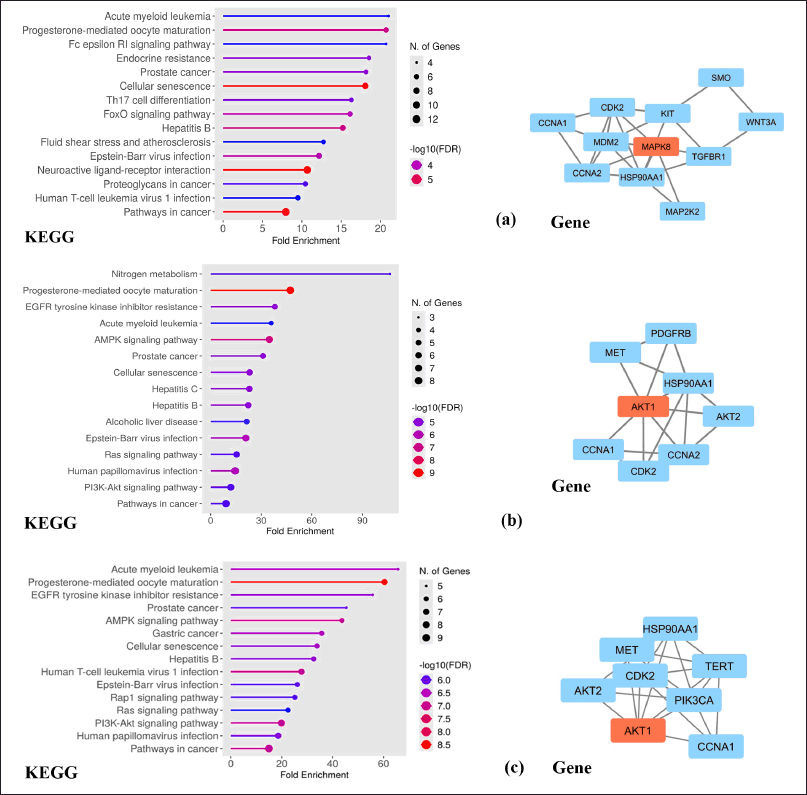 | Figure 5. Top 15 network pharmacology pathways based on KEGG, and determination of key proteins involved pathways in cancer for compound (a) A, (b) B, and (c) C. [Click here to view] |
The main target protein for compound A is MAPK8, which is also known as c-JUN N-terminal kinase found in glioblastoma cells. Induction of this protein will undergo cellular transformation and affect epidermal growth and platelet decline, as well as a signaling pathway responsible for cell adhesion and migration in breast cancer cells [35]. The activation of MAPK8 is closely related to chemotherapy resistance for paclitaxel, mechlorethamine, and doxorubicin by reducing caspase activation, inhibiting microtubules, and supporting DNA fragmentation after breast cancer treatment with alkylating agents and anthracyclines [36]. Other reported studies stated that suppression of MAPK8 protein expression plays a major role in preventing the proliferation process of breast cancer cells [37]. The ER is closely related to breast cancer and interacts with membrane receptors and key signaling molecules that activate the MAPK and phosphatidylinositol-3 kinase (PI3K)/AKT pathways. Thus, inhibition of estrogen pathway signaling becomes one of the effective methods in the treatment of breast cancer [38].
The main protein of compounds B and C is AKT1 as revealed from the bioinformatic analysis of pathways in cancer network pharmacology. AKT1 protein is a key element of the PI3K/AKT signaling pathway. AKT1 regulates cancer characteristics such as tumor growth, survival, tumor cell invasion, proliferation, apoptosis, and angiogenesis [39]. AKT is controlled by PI3K as the main regulator of activation and is an important part that has been validated as a promising therapeutic target [40]. AKT1 is expressed in most tissues, and the mutations could activate the PI3K/AKT pathway and eliminate phosphatase and tensin homologue [41], involved in proliferation and growth, promote tumor initiation, and suppress apoptosis [42].
Molecular docking analysis
Molecular docking studies of each compound were performed on each main protein obtained from the bioinformatics data. Molecular docking studies of curcumin analogs (Fig. 6) for compound A were carried out with MAPK8 (PDB ID 2G01) [43] and compounds B and C were carried out with AKT1 (PDB ID: 4EJN) [44]. Compound A has a binding affinity energy of −9.4 kcal/mol and an RMSD value of 0.71 ?. The observed interactions with amino acid residues are van der Waals interaction (Glu109, Ser155, and Val186); alkyl and pi-alkyl (Ala36, Val40, Ala53, Leu110, Met111, Val158, Leu168, and Val187); and pi-sulfur (Lys55, Met108). Meanwhile, the native ligand has a binding affinity energy of −7.6 kcal/mol and an RMSD value of 1.83 ? with hydrogen bond (Met111, Asn114), alkyl, and pi-alkyl (Ile32, Val40, Ala53, Lys55, Leu110, Val158, and Leu168), and pi-sigma (Val158) interactions. The stability of the intermolecular bonds with the protein receptor shows that compound A has better stability compared to the original ligand. In contrast, the therapeutic activity against breast cancer of compound A has not shown better activity. Compound B has a binding affinity energy of −10.5 kcal/mol and an RMSD value of 1.03 ?. The type of interaction with amino acid residues is hydrogen bond interaction (Gln79, Thr82, and Ser205), as well as alkyl and pi-alkyl (Trp80, Ile84, Leu210, Leu264, Lys268, Arg273, Cys296, and Cys310). Compound C has binding affinity energy of −10.7 kcal/mol, an RMSD value of 0.71 ?, and various interaction types via hydrogen bonding (Ser205), van der Waals (Gln203), alkyl, and pi-alkyl (Trp80, Ile84, Leu210, Leu264, Lys268, Arg273, and Cys310), and pi-anion (Asp274). Besides, both compounds B and C give lower binding affinity energy than native ligands (−13.6 kcal/mol, RMSD value 1.37 ?). This is caused by the native ligand exhibit more interactions with amino acid residues, i.e., hydrogen bond interactions (Ser56, Leu78, Thr83, and Gln203), van der Waals (Gln59), alkyl, and pi-alkyl (Ala58, Trp80, Leu264, Lys268, and Val270), pi-pi stacked (Trp80), pi-sigma (Lys268), and halogen bond (Asn53). The bond stability between the evaluated compounds to protein receptors shows that the order of better bond stability starts from the native ligand, compound C, and compound B. The IC50 results from the in vitro activity test of each curcumin analog are supported by bioinformatic data on network pharmacology in cancer pathways. However, the molecular docking studies do not match well as they have not provided a correct activity order. Because of that, further development is needed to obtain curcumin analogs with better activity and bond stability to optimize breast cancer therapy in the future.
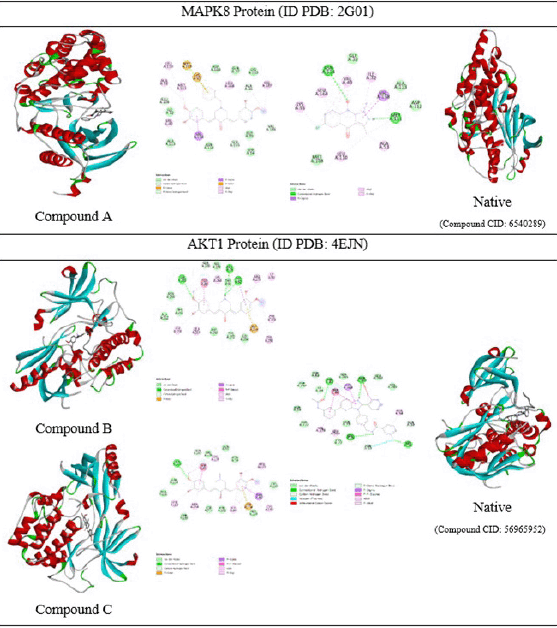 | Figure 6. 3D and 2D molecular docking visualizations of three curcumin analogs against protein receptors along with native ligands. [Click here to view] |
CONCLUSION
Synthesis and pharmacokinetic evaluation of curcumin analogs A, B, and C showed improvements in absorption, tissue distribution, retention, and safer toxicity profiles compared to curcumin. The identified main limitation was in oral bioavailability due to non-compliance with Lipinski’s rule of five. The anticancer activity tests against T47D cells showed IC50 values of 43.04, 19.20, and 20.00 µg/ml for compounds A, B, and C, respectively. Against HER2 cells, compound A gave IC50 value of 441.96 µg/ml, while compounds B and C gave IC50 values of 30.70 and 87.63 µg/ml. Compounds A and B gave IC50 values of 277.72 and 564.17 µg/ml, while compound C gave IC50 value of 26.63 µg/ml against MCF-7 cells. For 4T1 cells, compounds A and B gave IC50 values of 174.05 and 101.55 µg/ml, while compound C gave IC50 value of 203.66 µg/ml. All compounds demonstrate low selectivity, as shown by their SI values < 1, indicating potential cytotoxic effects on normal cells that may limit clinical utility. Bioinformatic studies of network pharmacology with pathways in cancer indicate MAPK8 protein as the primary target of compound A while AKT1 protein becomes the primary target for compounds B and C analogs. Future research on targeted delivery strategies and structural modifications is recommended to improve selectivity and optimize the efficacy and safety of these compounds for therapeutic applications.
ACKNOWLEDGMENT
The authors would like to thank Universitas Gadjah Mada for providing the research funding through the Academic Excellence Improvement Program Scheme B in 2024 (No. 4416/UN1/DITLIT/PT.01.03/2024), as well as supporting facilities from the Chemistry Study Program, Faculty of Mathematics and Natural Sciences, Universitas Gadjah Mada.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to the conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
CONFLICT OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included in this research article.
PUBLISHER’S NOTE
All claims expressed in this article are solely those of the authors and do not necessarily represent those of the publisher, the editors and the reviewers. This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
USE OF ARTIFICIAL INTELLIGENCE (AI)-ASSISTED TECHNOLOGY
The authors declares that they have not used artificial intelligence (AI)-tools for writing and editing of the manuscript, and no images were manipulated using AI.
REFERENCES
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71:209–49. CrossRef
2. Thakur C, Qiu Y, Fu Y, Bi Z, Zhang W, Ji H, et al. Epigenetics and environment in breast cancer: new paradigms for anti-cancer therapies. Front Oncol. 2022;12:971288. CrossRef
3. Novitasari D, Jenie RI, Utomo RY, Kato JY, Meiyanto E. CCA-1.1, a novel curcumin analog, exerts cytotoxic anti-migratory activity toward TNBC and HER2-enriched breast cancer cells. Asian Pac J Cancer Prev. 2021;22:1827–36. CrossRef
4. Burguin A, Diorio C, Durocher F. Breast cancer treatments: updates and new challenges. J Pers Med. 2021;11:808. CrossRef
5. Stoicescu EA, Iancu RC, Cherecheanu AP, Iancu G. Ocular adverse effects of anti-cancer chemotherapy. J Med Life. 2023;16:818–21. CrossRef
6. Khudhayer Oglah M, Fakri Mustafa Y. Curcumin analogs: synthesis and biological activities. Med Chem Res. 2020;29:479–86. CrossRef
7. Wolosewicz K, Podgorska K, Rutkowska E, Lazny R. Synthesis of Dicarbonyl curcumin analogues containing the tropane scaffold. European J Org Chem. 2019;2019:4662–74. CrossRef
8. Ardiansah B, Hardhani MR, Putera DDSR, Wukirsari T, Cahyana AH, Jia JW, et al. Design, synthesis, and antioxidant evaluation of monocarbonyl curcumin analogues tethered 1,2,3-triazole scaffold. Case Stud Chem Environ Eng. 2023;8:100425. CrossRef
9. Hani U, Jaswanth Gowda BH, Siddiqua A, Wahab S, Begum Y, Sathishbabu P, et al. Herbal approach for treatment of cancer using curcumin as an anticancer agent: a review on novel drug delivery systems. J Mol Liq. 2023;390:123037. CrossRef
10. Kunnumakkara AB, Bordoloi D, Padmavathi G, Monisha J, Roy NK, Prasad S, et al. Curcumin, the golden nutraceutical: multitargeting for multiple chronic diseases. Br J Pharmacol. 2017;174:1325–48. CrossRef
11. Al-Mulla A. A review: biological importance of heterocyclic compounds. Der Pharma Chemica. 2017;9:141–7.
12. Ansari A, Ali A, Asif M, Shamsuzzaman. Review: biologically active pyrazole derivatives. New J Chem. 2016;41:16–41. CrossRef
13. Al-Hujaily EM, Mohamed AG, Al-Sharif I, Youssef KM, Manogaran PS, Al-Otaibi B, et al. PAC, a novel curcumin analogue, has anti-breast cancer properties with higher efficiency on ER-negative cells. Breast Cancer Res Treat. 2011;128:97–107. CrossRef
14. Adams BK, Cai J, Armstrong J, Herold M, Lu YJ, Sun A, et al. EF24, a novel synthetic curcumin analog, induces apoptosis in cancer cells via a redox-dependent mechanism. Anticancer Drugs. 2005;16:263–75. CrossRef
15. Yuan X, Li H, Bai H, Su Z, Xiang Q, Wang C, et al. Synthesis of novel curcumin analogues for inhibition of 11β-hydroxysteroid dehydrogenase type 1 with anti-diabetic properties. Eur J Med Chem. 2014;77:223–30. CrossRef
16. Gurning K, Suratno S, Astuti E, Haryadi W. Untargeted LC/HRMS metabolomics analysis and anticancer activity assay on MCF-7 and A549 cells from coleus amboinicus lour leaf extract. IJ Pharm Res. 2024;23:e143494. CrossRef
17. Islamie R, Iksen I, Buana BC, Gurning K, Syahputra HD, Winata HS. Construction of network pharmacology-based approach and potential mechanism from major components of Coriander sativum L. against COVID-19. Pharmacia. 2022;69:689–97. CrossRef
18. Haryadi W, Gurning K, Astuti E. Molecular target identification of two Coleus amboinicus leaf isolates toward lung cancer using a bioinformatic approach and molecular docking-based assessment. J Appl Pharm Sci. 2024;14:203–10. CrossRef
19. Haryadi W, Gurning K, Fachiroh J, Astuti E. Potential of bioactive compounds in Coleus amboinicus, Lour., leaves against breast cancer by assessment using a network pharmacology approach and cytotoxic test. J Multidisciplinary Appl Nat Sci. 2025;5:267–87. CrossRef
20. Liu J, Liu J, Tong X, Peng W, Wei S, Sun T, et al. Network pharmacology prediction and molecular docking-based strategy to discover the potential pharmacological mechanism of huai hua san against ulcerative colitis. Drug Des Devel Ther. 2021;15:3255–76. CrossRef
21. Haryadi W, Pranowo HD. Molecular docking and dynamics analysis of halogenated imidazole chalcone as anticancer compounds. Pharmacia. 2023;70:323–9. CrossRef
22. Bitew M, Desalegn T, Demissie TB, Belayneh A, Endale M, Eswaramoorthy R. Pharmacokinetics and drug-likeness of antidiabetic flavonoids: Molecular docking and DFT study. PLoS One. 2021;16:e0260853. CrossRef
23. Hou T, Wang J, Zhang W, Xu X. ADME evaluation in drug discovery. 7. prediction of oral absorption by correlation and classification. J Chem Inf Model. 2007;47:e0260853. CrossRef
24. Smith DA, Beaumont K, Maurer TS, Di L. Volume of distribution in drug design. J Med Chem. 2015;58:5691–8. CrossRef
25. Zanger UM, Schwab M. Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol Ther. 2013;138:103–41. CrossRef
26. Bertholee D, Maring JG, van Kuilenburg ABP. Genotypes affecting the pharmacokinetics of anticancer drugs. Clin Pharmacokinet. 2017;56:317–37. CrossRef
27. Maurya R, Vikal A, Patel P, Narang RK, Kurmi B Das. “Enhancing oral drug absorption: overcoming physiological and pharmaceutical barriers for improved bioavailability.” AAPS PharmSciTech. 2024;25:228. CrossRef
28. Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. CrossRef
29. Liu Q, Liu N, van der Noord V, van der Stel W, van de Water B, Danen EHJ, et al. Differential response of luminal and basal breast cancer cells to acute and chronic hypoxia. Breast Cancer Res Treat. 2023;198:583–96. CrossRef
30. Rabinovich I, De Noronha L, Sebastião APM, Lima RS, Urban CA, Schunemann E, et al. HER2-expressing breast tumors are associated with breast cancer stem-cell phenotype CD44 + /CD24. J Bras Patol Med Lab. 2018;54:310–8. CrossRef
31. Arroyo-Crespo JJ, Armiñán A, Charbonnier D, Deladriere C, Palomino-Schätzlein M, Lamas-Domingo R, et al. Characterization of triple-negative breast cancer preclinical models provides functional evidence of metastatic progression. Int J Cancer. 2019;145:2267–81. CrossRef
32. Fernandes TB, Damião MFCB, Polli MC, Filho RP. Analysis of the applicability and use of Lipinski`s rule for central nervous system drugs. Lett Drug Des Discov. 2016;13:999–1006. CrossRef
33. Ugwu DI, Conradie J. Anticancer properties of complexes derived from bidentate ligands. J Inorg Biochem. 2023;246:112268. CrossRef
34. Romano JD, Tatonetti NP. Informatics and computational methods in natural product drug discovery: a review and perspectives. Front Genet. 2019;10:442506. CrossRef
35. Xu P, Zhang G, Hou S, Sha L gui. MAPK8 mediates resistance to temozolomide and apoptosis of glioblastoma cells through MAPK signaling pathway. Biomed Pharmacother. 2018;106:1419–27. CrossRef
36. Marinello PC, Panis C, Silva TNX, Binato R, Abdelhay E, Rodrigues JA, et al. Metformin prevention of doxorubicin resistance in MCF-7 and MDA-MB-231 involves oxidative stress generation and modulation of cell adaptation genes. Sci Rep. 2019;9:1–11. CrossRef
37. Wang R, Zhao Y. Effects of metformin on JNK signaling pathway and PD-L1 expression in triple negative breast cancer. Cancer Manag Res. 2024;16:259–68. CrossRef
38. Zhang YH, Zeng T, Chen L, Huang T, Cai YD. Determining protein–protein functional associations by functional rules based on gene ontology and KEGG pathway. Biochimica et Biophysica Acta (BBA)—Proteins Proteomics. 2021;1869:140621. CrossRef
39. Hinz N, Jücker M. Distinct functions of AKT isoforms in breast cancer: a comprehensive review. Cell Commun Signaling. 2019;17:1–29. CrossRef
40. Shariati M, Meric-Bernstam F. Targeting AKT for cancer therapy. Expert Opin Investig Drugs. 2019;28:977–88. CrossRef
41. Miricescu D, Totan A, Stanescu-Spinu II, Badoiu SC, Stefani C, Greabu M. PI3K/AKT/mTOR Signaling pathway in breast cancer: from molecular landscape to clinical aspects. Int J Mol Sci. 2020;22:173. CrossRef
42. Martorana F, Motta G, Pavone G, Motta L, Stella S, Vitale SR, et al. AKT Inhibitors: new weapons in the fight against breast cancer? Front Pharmacol. 2021;12:662232. CrossRef
43. Srinivasan M, Gangurde A, Chandane AY, Tagalpallewar A, Pawar A, Baheti AM. Integrating network pharmacology and in silico analysis deciphers Withaferin-A’s anti- breast cancer potential via hedgehog pathway and target network interplay. Brief Bioinform. 2024;25:bbae032. CrossRef
44. Khzem AH Al, Alturki MS, Almuzaini OK, Wali SM, Almaghrabi M, Aldawsari MF, et al. Isoetin from Isoetaceae exhibits superior pentatransferase inhibition in breast cancer: comparative computational profiling with FDA-approved Tucatinib. Pharmaceuticals. 2025;18:662. CrossRef