
INTRODUCTION
Bruton’s tyrosine kinase (BTK) is identified as a member of the Tec family kinase, as established by Smith et al. [1] in 2001 and Gomez-Rodriguez et al. [2] in 2007. Its prominent expression in hematopoietic cells and classification as one of the largest cytoplasmic kinases have been documented by Smith et al. [3], Genevier et al. [4] in 1994, and Mohamed et al. [5] in 2009. BTK, according to the findings of Kurosaki [6] in 1997, Mahajan et al. [7] in 1999, and Mao et al. [8] in 2001, plays a pivotal role in multiple signal transduction pathways governing the activation, development, differentiation, growth, proliferation, and survival of B lymphoid cells. The activation of BTK, in conjunction with several essential transcription factors, is orchestrated through a cascade initiated by an active B-cell receptor. This activation proves indispensable for B-cell proliferation and differentiation, as supported by the research of Petro et al. [9] in 2000 and Ponader et al. [10] in 2012. Notably, the activation of BTK by Src-family kinases sets off downstream activation of fundamental cell survival pathways, including MAP-Kinase and NFB, as observed by Herman et al. [11] in 2011.
X-linked agammaglobulinemia (XLA) is a condition associated with mutations in the BTK gene. In males, XLA leads to a deficiency in mature B-cells and serum albumin, resulting in decreased levels of immunoglobulins in the serum and susceptibility to persistent infections, as reported by Tsukada and Witte [12] in 1994. Given that rheumatoid arthritis (RA) is characterized by polyclonal B-cell activation and B-cell growth, the selective suppression of BTK presents an attractive treatment approach for both RA and B-cell lymphoma. BTK’s unique therapeutic potential stems from its selectivity for B-cells and its nonlethal nature, as outlined by Herman et al. [11] in 2011. In a previous study, we employed an extensive ligand-based virtual screening approach to identify prospective novel inhibitors, including ZINC85569481, BE015, ZINC08790868, and BE009, as highlighted in Sakthivel and Habeeb [13] in 2018. The use of QSAR as a tool has proven effective and promising in predicting molecular activity and elucidating chemo-biological interactions related to the studied biological activity, as discussed by Ghodsi and Hemmateenejad [14] in 2016.
However, a previous study conducted by Ali et al. [15] in 2021, which reported the potential of imidazo [1,5-a] pyrazine derivatives, relied solely on atom-based QSAR models, providing only rough approximations. Previous studies using different alignment strategies in QSAR modeling have demonstrated effective predictive power [3].Therefore, a more accurate approach for lead discovery and optimization can be achieved by both field-based and Gaussian-based 3D-QSAR models.
In another study, Ahmadi et al. [17] in 2021 utilized a hybrid descriptor-based approach involving Monte–Carlo methods to model BTK inhibitors. In our present study, we explored a 3D-QSAR analysis on a series of derivatives of 8-amino-imidazo [1, 5-a] pyrazines and optimized CGI-1746 derivatives, which were evaluated for their potential to inhibit BTK, as documented by Liu et al. [18] and Young et al. [19] in 2016.
MATERIALS AND METHOD
Dataset
A set of 66 potential inhibitors of BTK showing in-vitro activity was collected after a thorough literature review [18,19]. The experimental IC50 value of inhibitors was converted to pIC50 [20]. Chemsketch software was used to create a visual representation of the 2D structure of 66 BTK inhibitors [21]. The ligands were created using the Ligprep module of Maestro 10.2 version. This included creating possible tautomers and stereospecificity based on the chiral centers contained in the ligands, converting their 2D structures into 3D structures, and optimizing them using the OPLS 2005 force field. Finally, a low-energy ring conformation was produced for every ligand [22]. Table 1 provides the experimental and anticipated IC50 values for every drug that was developed using Schrodinger 10.2 to create 3D-QSAR models, which were based on Field and Gaussian techniques.
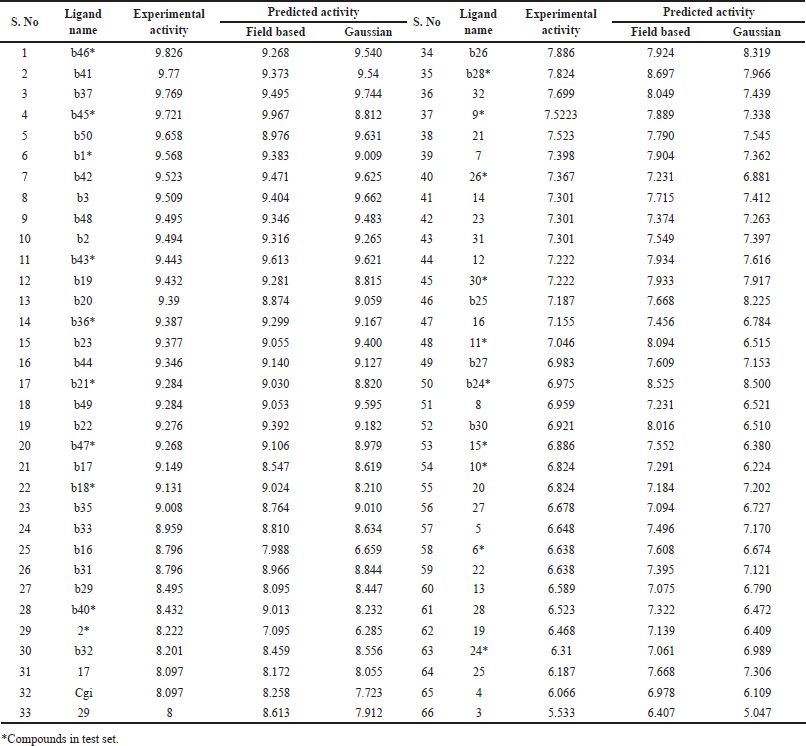 | Table 1. Experimental and QSAR predicted activity of compounds. [Click here to view] |
Alignment and protein preparation
The focus of this study is on developing field-based 3D-QSAR models, where the accurate alignment of compounds is crucial. For this purpose, the molecular docking process to establish a receptor-based alignment was enforced by downloading the experimental structure of BTK (PDB ID: 3OCS). To confirm that the structure was ready for analysis, Schrodinger 10.2v’s protein preparation wizard was implemented to remove water molecules followed by the optimization of H-bond configurations. Careful and close attention was also paid to optimizing the target protein and OPLS 2005 force field-based minimization (heavy atom convergence = 0.5 A°) [23].
Receptor grid generation and docking
The receptor grid generating module in Schrodinger 10.2v was utilized to build the receptor grid used in the current study. The spatial organization of the receptors that are explored during the docking process is provided by this grid file. The residues around the active site, which includes LEU542, THR410, GLY411, THR474, LEU528, ALA428, TYR476, ALA479, ALA478, GLY480, SER453, ASN526, ASP459, GLU475, VAL416, and LEU408, were particularly included in the grid that was created [24]. The Glide XP technique was then used to dock the known inhibitors to BTK.
Field-based and Gaussian-based 3D-QSAR model calculations
The 3D-QSAR analysis in Schrödinger 10.2v employed both field-based and Gaussian-based methods, following the approach outlined by Myint and Xie [25] and Verma et al. [26]. The methodology involved establishing a 3D-QSAR model that correlates the 3D characteristics with the activities of aligned molecules. Construction of the QSAR model utilized partial least square (PLS) regression analysis, incorporating five factors. The independent variables for this analysis were the field and Gaussian intensities, computed based on the steric and electrostatic fields surrounding the ligand molecule within a 3D grid.
In the domain of multivariate statistical analysis, the field-based 3D-QSAR model operates as an alignment-dependent technique, establishing a relationship between molecular field interaction energy terms and biological activities. This model computes the electrostatic and steric fields within a grid surrounding the ligand molecule and subsequently correlates experimental pIC50 values with these interaction energy terms. Conversely, the Gaussian 3D-QSAR model incorporates hydrophobic, hydrogen bond donor (HBD), and hydrogen bond acceptor (HBA) fields, along with other potential fields, to calculate interaction energy. The determination of these potential fields relies on Gaussian equations. Both the field-based and Gaussian 3D-QSAR models share the common objective of generating predictive 3D-QSAR models. These models aim to forecast the biological activities of ligand molecules based on their interaction energies with the surrounding environment.
RESULTS AND DISCUSSION
Generation of field-based and Gaussian-based QSAR models
To gain a thorough understanding of the biological significance associated with each structural fragment and to establish a statistical relationship between activity and chemical attributes, we employed a receptor-based alignment method derived from Glide XP docking in our study. This method served as the basis for creating both field-based and Gaussian-based QSAR models. Figure 1A illustrates the positioning of inhibitors within the BTK binding site, while Figure 1B displays the alignment of ligands. The 3D-QSAR model was constructed using the most favorable docked configurations from Glide XP. We generated a field-based 3D-QSAR model with two key features, steric and electrostatic, and a Gaussian-based 3D-QSAR model with five features: Gaussian steric, Gaussian electrostatic, Gaussian hydrophobic, H-BA, and H-BD.
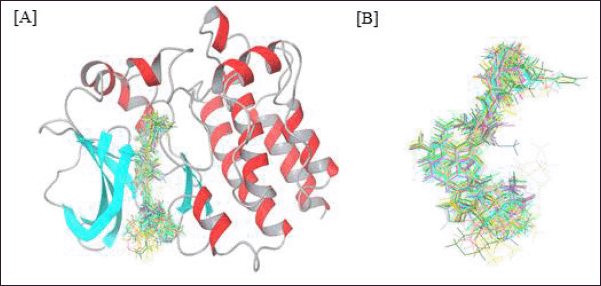 | Figure 1. (A) Alignment of known inhibitors in the binding site of BTK. (B) Ligand alignment after docking. [Click here to view] |
For model generation and validation, all compounds were randomly divided into training and test sets. PLS analysis was conducted with 46 compounds in the training set and 20 compounds in the test set, employing five PLS factors to produce a model with acceptable q2 and r2 values. The experimental and predicted IC50 values are provided in Table 1 for reference.
Field-based 3D-QSAR model
A force field-based 3D-QSAR model was created by establishing a correlation between the steric and electrostatic field intensities of the training set. The effectiveness of the model was assessed using various methods. A considerable degree of correlation was shown by the r2cv value of 0.6952, which was obtained using the Leave One Out (LOO) cross-validation method. Furthermore, a study conducted without cross-validation produced a r2 value of 0.9292, indicating a robust connection. Listed in Table 2, the standard error of 0.33 and F ratio of 110 provide further insights into the model’s accuracy and significance. The electrostatic and steric features of the compounds were analyzed using contour maps.
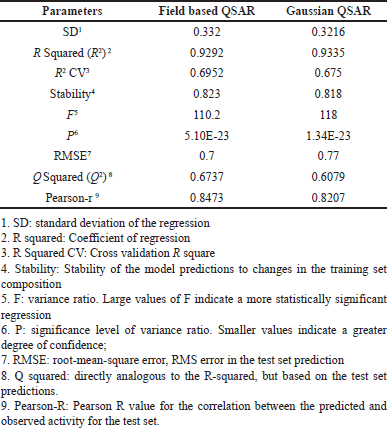 | Table 2. Statistical summary of the 3D QSAR model. [Click here to view] |
Steric contour maps
Steric contour maps visually depict favorable and unfavorable steric bulks. In Figure 2A and B, the green and yellow colors represent field-based steric interactions. To elaborate, the green regions within the molecule signify that the introduction of bulky substituents has the potential to augment its activity. Conversely, the yellow regions indicate that the inclusion of bulky components may diminish the molecule’s activity. For instance, when examining the highly active molecule b46 (Fig. 2A), the presence of the green color in the benzene ring implies that incorporating bulky substitutions at this location could potentially enhance the molecule’s activity. In the molecule with the lowest activity, molecule 3 (Fig. 2B), the presence of a green contour near C5 of the benzene molecule suggests that the addition of a bulky group at that position could potentially enhance the molecule’s activity. The steric field’s contribution to this observation was measured to be 0.73.
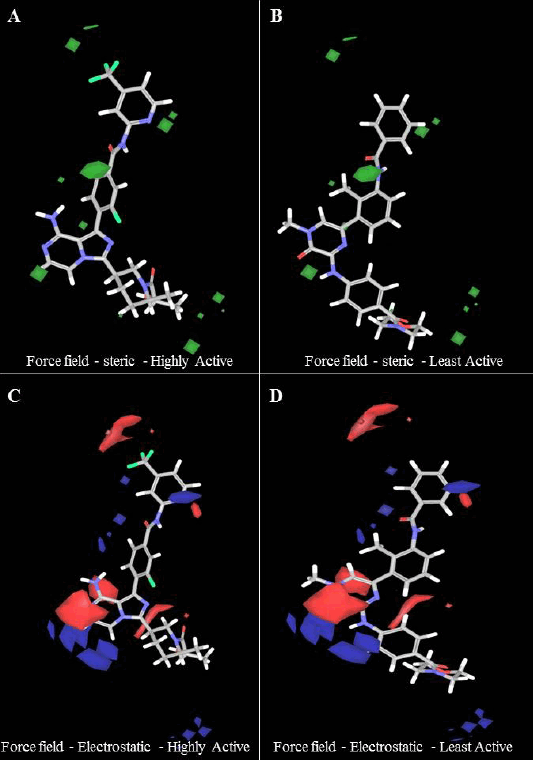 | Figure 2. (A and B) Steric contour and (C and D) electrostatic contour map for the best field-based 3D – QSAR model. For the steric model, favorable regions were shown in green, and in the electrostatic contour map, favorable electropositive regions were shown in blue, and electronegative regions were shown in red color. [Click here to view] |
Electrostatic contour maps
In Figure 2C and 2D, field-based electrostatic interactions for the most reactive and least reactive molecules are depicted. The highly reactive molecule is represented by blue color, indicating electropositive regions, which have the potential to enhance its activity. On the other hand, the least reactive molecule is shown in red, signifying electronegative regions, suggesting that the presence of electronegative groups may increase or enhance its activity. It is worth noting that the contribution of the electrostatic force field was 0.27.
Steric and electrostatic contour maps for the highest, moderate, and lowest active compounds are shown in supplementary Figure S1. The combined electrostatic contour map unmistakably shows that incorporating both electropositive and electronegative groups will enhance the biological activity of the compounds.
Gaussian-based 3D-QSAR model
Table 2 presents the statistical details of the Gaussian-based QSAR model, encompassing the electrostatic, HBA, HBD, hydrophobic, and steric fields, along with their corresponding field contributions. The model’s performance underwent evaluation through various methods. The LOO cross-validation method yielded an r2cv value of 0.675, indicating a moderate level of correlation. Furthermore, a non-cross-validation analysis resulted in an r2 value of 0.9335, suggesting a robust correlation between the model’s predictions and the actual data. The standard error of the estimate was measured at 0.32, and the F ratio was calculated to be 118, underscoring both the accuracy and significance of the model.
The contributions of the steric, electrostatic, hydrophobic, HBA, and HBD fields were 0.385, 0.099, 0.293, 0.161, and 0.063, respectively. In comparison, the steric (0.385) and hydrophobic (0.293) field intensities exhibited higher contributions than the electrostatic, HBA, and HBD fields. This observation implies that in protein-ligand interactions, there is a greater emphasis on steric and hydrophobic fields, underscoring their significance in influencing the biological activity of compounds.
Gaussian steric contour maps
Figure 3A and 3a depicts the Gaussian steric contour maps for the highly active molecule (B46) and the least active molecule (compound 3). The green regions in the molecule indicate positions where the presence of bulky substituents is favorable and likely to enhance the activity. Conversely, the yellow regions suggest that incorporating bulky groups at these positions may diminish the activity. Notably, in the case of the least active molecule, it is evident that the introduction of bulky substitutions (indicated by the green color) near the first benzene ring of both B46 and compound 3 may enhance the compound’s activity. The Gaussian steric contribution was determined to be 0.385, and this value was higher compared to the contributions of other factors, underscoring its significant influence on the compound’s biological activity.
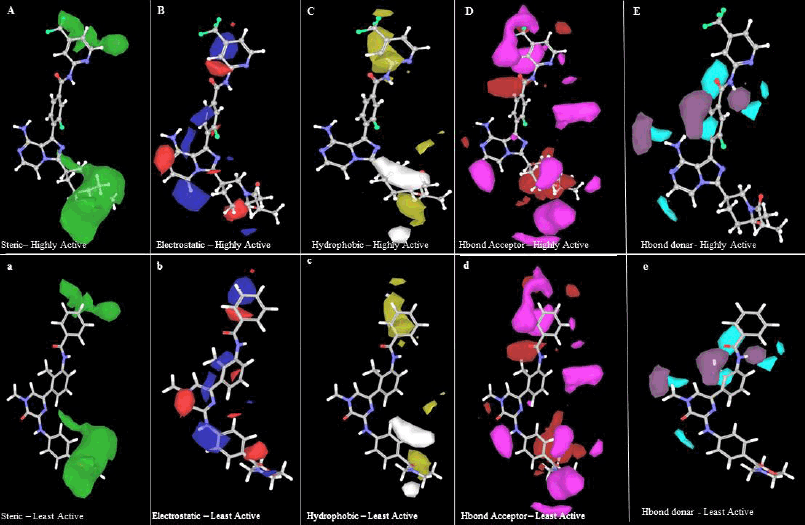 | Figure 3. Gaussian steric contour maps for (A) most active (B46) and (a) least active (compound 3); Gaussian electrostatic contour maps for (B) most active (B46) and (b) least active (compound 3); Gaussian hydrophobic contour maps for (C) most active (B46) and (c) least active (compound 3); Gaussian HBA contour maps for (D) most active (B46) and (d) least active (compound 3); Gaussian HBD contour maps for (E) most active (B46) and (e) least active (compound 3). [Click here to view] |
Gaussian electrostatic contour maps
The Gaussian electrostatic contour maps for the highly active molecule (B46) and the least active molecule (compound 3) are displayed in Figure 3B and 3b, respectively. In these figures, the blue and red contours symbolize electropositive and electronegative groups, respectively, which hold the potential to augment the biological activity of the compounds. The electrostatic contribution was determined to be 0.161, ranking as the second lowest when compared to other contributions. In case of the most active molecule, the presence of a large red contour map around the hydrindane and a blue contour map at the benzene and methyl groups suggests that electronegative and electropositive atoms at these positions may positively influence the compound’s activity, enhancing it. For the least active molecule, it is noteworthy that the addition of an electronegative group near the C4 methyl group of the first benzene ring could potentially increase the compound’s activity.
Hydrophobic
In Figure 3C and 3c, the yellow color (hydrophobic groups) at that position is tolerated and the white contour represents unfavored hydrophobic groups. In the highest active molecule, the yellow contour near methyl benzene shows good activity. The white contour represents in second benzene ring represents the disfavored hydrophobic group. The overall contribution of hydrophobic in our model is 0.293.
HBA and HBD
In Figure 3D and 3d, an HBA contour map for the most and least active molecules is presented. The red contour in the HBA indicates improved activity, while the magenta contour suggests that the activity may be reduced due to the presence of HBA groups. Specifically, in compound 3, a red contour near the oxygen atom of methoxybenzene is identified to favor the activity of the compound. This oxygen atom accepts electrons from LYS430 to form hydrogen bonds. Figure 3E and 3e represent the HBD contour. The purple contour represents the area favored by donors and unpreferred areas are shown in cyan color. The least activity of compound 3 may be due to the acceptor and donor (highlighted) forming a hydrogen bond with MET477. The contribution of HBA and HBD in the build model was 0.161 and 0.063, respectively.
Validation of field- and Gaussian-based 3D-QSAR models
The scatter plots, comparing predicted versus experimental activity for the training and test set inhibitors, are depicted in Figure 4 for the field-based 3D-QSAR model and in Figure 5 for the Gaussian-based 3D-QSAR model. These plots offer a visual representation of the model’s performance in predicting the biological activity of the compounds.
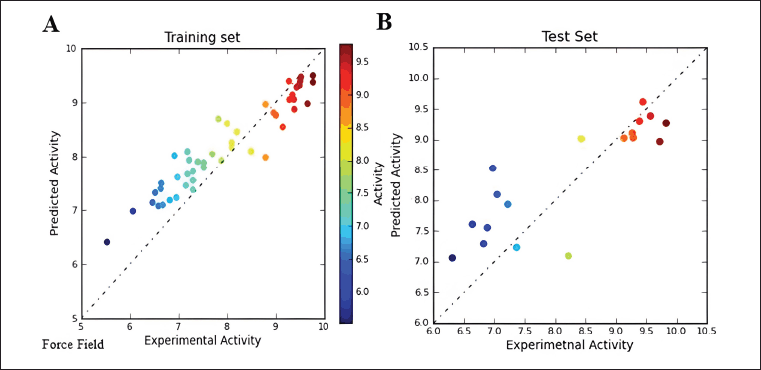 | Figure 4. Force field Scatter plot between observed and predicted activity of (A) training set and (B) test set compounds. [Click here to view] |
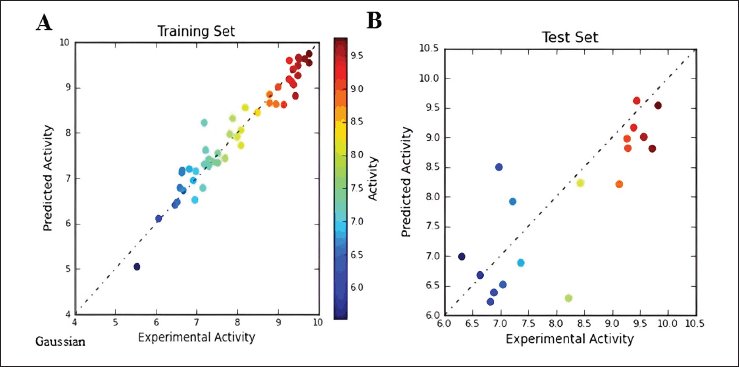 | Figure 5. Gaussian Scatter plot between observed and predicted activity of (A) training set and (B) test set compounds. [Click here to view] |
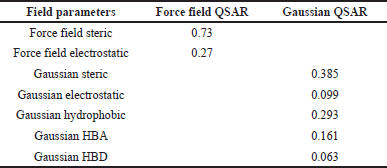 | Table 3. Field contribution of force field and Gaussian QSAR. [Click here to view] |
Gaussian Scatter plot between observed and predicted activity are provided in Supplementary Figure S2. The correlation coefficients (r2) for the field-based model and Gaussian model are 0.9292 and 0.9335, respectively. The difference in r2 values between the field-based and Gaussian models is not significant. The comparison between experimentally observed and predicted IC50 values reveals the Gaussian model’s effectiveness in predicting the activities of both the training and test molecules. The statistical analysis underscores that both models exhibit robust predictive capabilities. Furthermore, they offer valuable insights into the chemical characteristics of the ligands, which can be instrumental in the development of inhibitors targeting BTK.
CONCLUSION
In conclusion, our study successfully developed robust 3D-QSAR models for BTK inhibitors, employing both field-based and Gaussian-based approaches. The field-based QSAR model, considering steric and electrostatic fields, demonstrated strong predictive power, reflected in an r2 value of 0.92 and a q2 value of 0.69. In parallel, the Gaussian-based model, incorporating five field intensities (Gaussian steric, Gaussian electrostatic, Gaussian H-BA, Gaussian H-BD, and Gaussian hydrophobic), exhibited outstanding performance, boasting an r2 value of 0.93 and a q2 value of 0.67. These models provide valuable insights for the rational design of novel compounds with potent BTK inhibitory activity.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
FINANCIAL SUPPORT
The authors would like to thank SRM Institute of Science and Technology, Kattankulathur for providing all the necessary support and motivation to execute this study. We also thank International Conference on New Horizons in Bioengineering: Fostering Academia-Industry Partnership (ICB-2024) for supporting this article.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included in this research article.
PUBLISHER’S NOTE
All claims expressed in this article are solely those of the authors and do not necessarily represent those of the publisher, the editors and the reviewers. This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
USE OF ARTIFICIAL INTELLIGENCE (AI)-ASSISTED TECHNOLOGY
The authors declares that they have not used artificial intelligence (AI)-tools for writing and editing of the manuscript, and no images were manipulated using AI.
REFERENCES
1. Smith CE, Islam TC, Mattsson PT, Mohamed AJ, Nore BF, Vihinen M. The Tec family of cytoplasmic tyrosine kinases: mammalian Btk, Bmx, Itk, Tec, Txk and homologs in other species. Bioessays. 2001;23:436–46. CrossRef
2. Gomez-Rodriguez J, Readinger JA, Viorritto IC, Mueller KL, Houghtling RA, Schwartzberg PL. Tec kinases, actin, and cell adhesion. Immunol Rev. 2007;218:45–64. CrossRef
3. Smith CI, Baskin B, Humire-Greiff P, Zhou JN, Olsson PG, Maniar HS, et al. Expression of Bruton’s agammaglobulinemia tyrosine kinase gene, BTK, is selectively down-regulated in T lymphocytes and plasma cells. J Immunol. 1994;152:557–65. CrossRef
4. Genevier HC, Hinshelwood S, Gaspar HB, Rigley KP, Brown D, Saeland S, et al. Expression of Bruton’s tyrosine kinase protein within the B cell lineage. Eur J Immunol. 1994;24:3100–5. CrossRef
5. Mohamed AJ, Yu L, Bäckesjö CM, Vargas L, Faryal R, Aints A, et al. Bruton’s tyrosine kinase (Btk): function, regulation, and transformation with special emphasis on the PH domain. Immunol Rev. 2009;228:58–73. CrossRef
6. Kurosaki T. Molecular mechanisms in B cell antigen receptor signaling. Curr Opin Immunol. 1997;9:309–18. CrossRef
7. Mahajan S, Ghosh S, Sudbeck EA, Zheng Y, Downs S, Hupke M, et al. Rational design and synthesis of a novel anti-leukemic agent targeting Bruton′ s tyrosine kinase (BTK), LFM-A13 [α-cyano-β-hydroxy-β-methyl-N-(2, 5-dibromophenyl) propenamide]. J Biol Chem. 1999;274:9587–99. CrossRef
8. Mao C, Zhou M, Uckun FM. Crystal structure of Bruton’s tyrosine kinase domain suggests a novel pathway for activation and provides insights into the molecular basis of X-linked agammaglobulinemia. J Biol Chem. 2001;276:41435–43. CrossRef
9. Petro JB, Rahman SJ, Ballard DW, Khan WN. Bruton’s tyrosine kinase is required for activation of IκB kinase and nuclear factor κB in response to B cell receptor engagement. J Exp Med. 2000;191:1745–54. CrossRef
10. Ponader S, Chen SS, Buggy JJ, Balakrishnan K, Gandhi V, Wierda WG, et al. The Bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood J Am Soc Hematol. 2012;119:1182–9. CrossRef
11. Herman SE, Gordon AL, Hertlein E, Ramanunni A, Zhang X, Jaglowski S, et al. Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765. Blood. 2011;117:6287–96. CrossRef
12. Tsukada S, Witte ON. X-linked agammaglobulinemia and Bruton’s tyrosine kinase. Adv Exp Med Biol. 1994;365:233–8. CrossRef
13. Sakthivel S, Habeeb SK. Combined pharmacophore, virtual screening and molecular dynamics studies to identify Bruton’s tyrosine kinase inhibitors. J Biomol Struct Dyn. 2018;36:4320–37. CrossRef
14. Ghodsi R, Hemmateenejad B. QSAR study of diarylalkylimidazole and diarylalkyltriazole aromatase inhibitors. Med Chem Res. 2016;25:834–42. CrossRef
15. Ali A, Ali A, Ahsan MJ. Discovery of structural prospects of Imidazo [1, 5-a] pyrazine derivatives as BTK inhibitors against cancer: a computational study. Lett Drug Des Discov. 2021;18:1165–77. CrossRef
16. Aparna V, Jeevan J, Ravi M, Desiraju GR, Gopalakrishnan B. 3D-QSAR studies on antitubercular thymidine monophosphate kinase inhibitors based on different alignment methods. Bioorg Med Chem Lett. 2006;16:1014–20. CrossRef
17. Ahmadi S, Lotfi S, Afshari S, Kumar P, Ghasemi E. CORAL: Monte Carlo based global QSAR modelling of Bruton tyrosine kinase inhibitors using hybrid descriptors. SAR QSAR Environ Res. 2021;32:1013–31. CrossRef
18. Liu J, Guiadeen D, Krikorian A, Gao X, Wang J, Boga SB, et al. Discovery of 8-amino-imidazo [1, 5-a] pyrazines as reversible BTK inhibitors for the treatment of rheumatoid arthritis. ACS Med Chem Lett. 2016;7:198–203. CrossRef
19. Young WB, Barbosa J, Blomgren P, Bremer MC, Crawford JJ, Dambach D, et al. Discovery of highly potent and selective Bruton’s tyrosine kinase inhibitors: pyridazinone analogs with improved metabolic stability. Bioorg Med Chem Lett. 2016;26:575–9. CrossRef
20. Selvaraj C, Tripathi SK, Reddy KK, Singh SK. Tool development for Prediction of pIC50 values from the IC50 values-A pIC50 value calculator. Curr Trend Biotechnol Pharm. 2011;5:1104–9.
21. Elyashberg M, Williams A. ACD/Structure elucidator: 20 years in the history of development. Molecules. 2021;26:6623. CrossRef
22. Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc. 1996;118:11225–36. CrossRef
23. Kalva S, Vinod D, Saleena LM. Field-and Gaussian-based 3D-QSAR studies on barbiturate analogs as MMP-9 inhibitors. Med Chem Res. 2013;22(11):5303–13. CrossRef
24. Laskowski RA. PDBsum: summaries and analyses of PDB structures. Nuc Acid Res. 2001;29:221–2. CrossRef
25. Myint KZ, Xie XQ. Recent advances in fragment-based QSAR and multi-dimensional QSAR methods. Int J Mol Sci. 2010;11:3846–66. CrossRef
26. Verma J, Khedkar VM, Coutinho EC. 3D-QSAR in drug design-a review. Curr Top Med Chem. 2010;10:95–115. CrossRef
SUPPLEMENTARY MATERIAL
The supplementary material can be accessed at the link here: [https://japsonline.com/admin/php/uploadss/4601_pdf.pdf]