
INTRODUCTION
Cancer is still the leading mortality cause, and in 2021, the National Center for Health Statistics reported 1,898,160 new cancer patients, of which 608,570 deaths have been reported [1] in the United States alone. Among cancers, non-small cell lung cancer (NSCLC) is identified to have the highest mortality and morbidity rate compared to colon, prostate, and breast cancer together [2]. Therefore, improving the standard of care for NSCLC patients is curtailed to identify new indications and therapeutic strategies, utilizing new research methodologies. One known predictive biomarker targeting NSCLC is epidermal growth factor receptors (EGFR), and the EGFR gene is located on chromosome 7p12-13. Mutations in EGFR genes lead to cancerous conditions [3]. EGFR belongs to the receptor tyrosine kinase (RTK) receptor family, which is responsible for the signal transduction cascade of the receptor and activates protein kinase (PK) [4]. PK is an essential regulatory factor for cell biology and is vital in the signal transduction cascade that regulates apoptosis, cell cycle progression, transcription, and immune response [5]. The main problem of EGFR mutation in NSCLC condition is with the RTK enzyme that phosphorylates the cellular protein to activate many other kinases or auto-activation. Thus, RTK functions as a switch on or off, governing many cellular functions. RTK is a subclass of protein kinases. RTK has the phosphate group attached to it. Phosphorylation of proteins by kinases leads to signal transduction within the cell and regulates cellular activity, such as cell division and cell proliferation [6]. In the case of NSCLC, one of the major clinical challenges lies in the abnormal auto-phosphorylation activity observed in RTKs, particularly EGFR. This auto-phosphorylation is an atypical phenomenon where the receptor becomes phosphorylated even in the absence of its natural activating ligand. As a result, the receptor undergoes continuous and prolonged dimerization, leading to persistent activation of downstream signaling pathways. This dysregulation interferes with normal apoptotic mechanisms and disrupts cellular signaling cascades, thereby contributing to unchecked cell proliferation and tumor progression. Due to these sustained conformational changes in the RTK domain, drugs designed to target these receptors, especially first-, second-, and third-generation EGFR tyrosine kinase inhibitors, tend to bind less effectively. The unstable binding affinity results in reduced therapeutic efficacy. Consequently, to maintain clinical benefit, patients often require higher doses over prolonged treatment durations. However, this escalated dosing regimen significantly increases the risk of developing adverse effects, such as drug resistance and hepatotoxicity. This limitation represents a major drawback of the currently available EGFR-targeting agents [7]. Therefore, overcoming the issue of drug resistance caused by conformational instability in mutant EGFR RTK receptors is crucial. Addressing this problem holds the potential to markedly improve the pharmacological management of NSCLC, thereby enhancing treatment outcomes and patient survival. Therefore, tackling the drug resistance due to EGFR RTK receptors will improve the pharmacotherapy for NSCLC or lung cancer.
Our current study targeted the allosteric region of the EGFR to cause conformational changes in the structure and biological activity of the whole protein. These conformational changes do not trigger the protein’s active site. The main reason is that it primarily binds non-covalently to form a stable protein−ligand complex [8], thus inhibiting prolonged auto-phosphorylation. Studies to identify the fourth-generation EGFR allosteric inhibitor against NSCLC led to the identification of ligand EA1045, an allosteric inhibitor that targets EGFR and prevents drug resistance in lung cancer [9]. EA1045 is a new chemical entity under phase 3 of clinical trials, and it is used to control the in silico screening of FDA-approved drugs. According to studies by Zhao et al. [10], compounds EAI001 and EA1045 irreversibly bind to the EGFR receptor and target the allosteric site. EAI045 has stronger binding to the EGFR allosteric site than EAI001. EA1045 interacts more closely with the allosteric site [10]. Hence, EAI001 and EA1045 are standard molecules for docking studies. EA1045 is a compound under investigation and shows dominant interaction with the allosteric site of EGFR. Therefore, exploring the allosteric site of the kinase family can provide a remedy for new pharmacotherapy of NSCLC [11]. A series of small molecules showing EGFR allosteric inhibition were synthesized by Caporuscio et al. [12]. Ten ligands had > or ≈50% at 50 μM allosteric inhibitory properties against EGFR have been reported. The 10 compounds can be utilized for in silico homology development, on which the FDA-approved drugs can be docked and evaluated for repurposing. Inhibition of the allosteric site blocks the kinase activity without displacing the ATP, stabilizing the whole kinase domain [13]. This has made a void in the available NSCLC therapy. Based on these findings, we would like to suggest a new potential fourth-generation EGFR inhibitor by in silico pharmacology methods using Schrodinger software.
MATERIALS AND METHODS
Computer simulation study
The computer simulation was carried out as per our previous publication [6]. The computer simulations study was performed in the Schrödinger® Maestro tool (www.schrodinger.com), which is a paid workstation with Ubuntu platform, Quadro P620/ PCle/SSE2 graphics card with Intel® Xenon® Gold 6130 CPU @ 2.10 GHz × 64 processors, and 134.8 GB RAM.
Ligand preparation
The Drug Bank database (www.drugbank.ca) with (?2800) FDA-approved drugs in SMILE format was downloaded and optimized using LigPrep [14]. The 3D coordinates for the molecules were generated. The ionization state of pH 7.5 was predicted [9], the tautomer of each molecule was generated, and the protein“ligand binding state was defined. Finally, these optimized molecules were minimized using the OPLS3e force field [15].
EGFR protein preparation
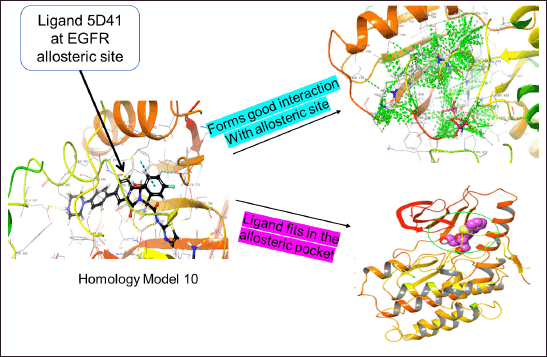
The protein structure with EGFR allosteric inhibitory ligands, namely EAI045 and EAI001, are extracted from the protein drug bank (PDB) (www.rcsb.org/) with codes 5ZWJ and 5D41. The protein structure was optimized from crystallography codes to computer-readable language using the protein preparation tool in Maestro, Schrödinger software [16]. The missing hydrogen molecules and amino-acid side chains were added to the protein−ligand complex, followed by the removal of bulky water molecules. Utilizing the PROPKA tool in Schrödinger software, the ionization state of the complex was set at pH 7.5 [17]. The minimized protein complex was obtained for the screening study. Protein−ligand interactions were done using a 2D visualizer in the Schrödinger tool. The allosteric ligands EAI045 and EAI001 showed hydrogen bond interaction and π−π stack activity with the protein. Multiple interactions were observed for both ligands with the proteins. Therefore, by docking studies, there is a chance to obtain high-affinity molecules targeting EGFR allosteric sites. The interaction sites in allosteric pockets were PHE 856, ASP 855, and LYS 745 (Fig. 1). However, the crystal structure was incomplete because of missing amino acid sequences. Hence, we went for homology model development and validation.
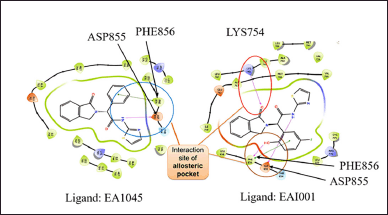 | Figure 1. 2D interaction diagram of protein-ligand interaction analysis of allosteric ligands EA1045 and EAI001. A hydrogen bond was formed with amino acid PHE856, ASP855, LYS754, and π-π stacking was formed through interactive forces with EAI001 at amino acid site PHE856 between the ligand-protein interaction. This indicates that the protein complex is stable during molecular docking. [Click here to view] |
Homology modeling
Since the 5D41 and 5ZWJ crystal structures were incomplete, the missing amino acid sequence of the respective proteins in the allosteric site was identified and extracted from www.uniport.org with reference UniProtKB-P00533. The homology modeling tool in Schrödinger software was used for this purpose. The incomplete protein structure was subjected to comparative analysis, where the template from the uniporter of the protein (the complete amino acid sequence), and the target proteins were optimized to complete the incomplete crystal structure [18]. The following steps were carried out: (i) identification of the amino acid sequence of each protein, (ii) identification of the related protein to obtain a comparative backbone of the target protein, (iii) sequence alignment of the amino acid sequence of the protein and template protein, (iv) building the missing loops in the protein by using Build tool in Schrodinger software, and (v) model refinement and evaluation by the BLAST tool in Schrodinger software.
The homology model of the respective protein was made using Schrödinger Prime software [19]. The model structure was later modified by refining the loop tool [20]. The pH was kept at 7.5 using PROPKA3. A restricted minimization technique in the Schrödinger tool further optimized the protein structure.
Selection of best ligands and homology structure for docking analysis
The allosteric ligands EA1045 and EAI001 were separated from the protein and underwent LigPrep. The receptor grid was generated at the allosteric pocket in the protein for all ten selected homology models using the PyMol tool in Schrodinger software. Ligand docking software from Schrödinger was used for visual analysis of the crystal structure. The extra precision (XP) docking method selected the best ligand and homology structure. The ligands were docked on the proteins with PDB IDs: 5D41 and 5ZWJ. Based on the interaction of ligand−protein, Ramachandran plot analysis and glide score analysis were performed to select the best two models for further analysis.
Few low-affinity molecules with IC50 value >10 µM were identified from the literature source of Caporuscio et al. [12]. The protein-ligand interaction is desired to be less than the glide score 5D41. The low-affinity molecules were sketched in 2D sketcher software, which is in-built in Schrödinger software. This 2D structure was converted into 3D and subjected to LigPrep. Grid was generated using the Glide tool in Schrödinger software in the allosteric site of homology models 1 and 10 (Table 1). The Ligand docking software of Schrödinger was used for visual screening analysis. The root mean square deviation (RMSD) helps identify and calculate the average changes in the complex of atoms in a molecule displaced from the network compared to the reference frame. All the trajectories in the RMSD calculation process were analyzed. Each frame is recorded for a particular time interval. This method for RMSD is repeated for all the generated frames in the simulation trajectory.
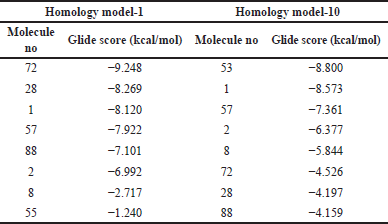 | Table 1. Glide score of homology models 1 and 10. [Click here to view] |
Molecular docking
The Glide module tool in Schrodinger software was used for the molecular docking study. The docking study helps to understand complex molecular interactions with the receptor system and optimized parameters to generate a broad spectrum of suitable ligand conformations and orientations within the target docking zone. In this study, key findings that were focused were hydrogen bonds, hydrophobic interactions, and pi−pi stacking interaction, which together will help to calculate the binding affinity [21]. Using the Receptor Grid Generation tool, a grid was generated to specify the allosteric region of the selected EGFR protein [22]. The optimized homology model was analyzed to identify five poses that showed the highest stability with maximum allosteric EGFR interaction between the ligand and protein. The five poses were identified by virtual screening and were named Protein A, Protein B, Protein C, Protein D, and Protein E, respectively. These five poses will be utilized for consensual docking analysis. Our study also aimed to compare consensual and normal docking; therefore, the process was carried out using ten homology models.
The US-FDA-approved molecules were subjected to LigPrep. The site scores of each model (Protein A, Protein B, Protein C, Protein D, and Protein E, respectively) were analyzed, and the pose with the highest site score was selected for each model individually. Grid was generated for each model separately, and the LigPrep molecules from FDA-approved drugs were docked for virtual screening analysis. First, a high-throughput visual screen (HTVS) was performed to identify the top 500 drugs. These 500 FDA drugs were docked in the grid generated by their respective models by the standard precision (SP) method. The top 100 drugs were identified and subjected to XP docking studies. The top 20 drugs were analyzed based on their protein−ligand interaction, GlideScore, and DockScore [23]. The homology model-1 was first docked on it for normal docking tops using the HTVS and SP methods. Further, the top 100 compounds showing good glide scores were analyzed by XP docking to identify the best drugs. In the study, two types of docking methodology were followed, namely consensual and normal docking, and further, the results were compared for the glide and dock score analysis to identify the top hit compound.
Molecular dynamic (MD) simulation study
The Schrodinger Desmond tool was used to conduct the MD simulation interaction study. The MD simulation study was done to predict the trajectory of the binding potential and configuration patterns of the protein−ligand complex. In silico, studies, MD analysis is the key tool used for validation of the stability of the protein−ligand complex. In addition, MD simulation analysis gives detailed information on the fluctuations and conformational changes of the protein−ligand complex toward its stable conformation [24]. We also analyzed the time-dependent protein−ligand interactions of the complex. A conformational dynamic of the complex system was analyzed. The homology structure number 10 of the protein 5D41, with the best stability and ligand interaction abilities, was observed during glide XP docking and further subjected to MD studies as per our previous work [25].
RESULTS
EA1045 and EAI001 ligand preparation
The ligands EA1045 and EAI001 showed structural similarity. The allosteric ligand EA1045 had an extra methyl group, showing strong hydrogen interaction with the target protein’s amino acid sequence. The IC50 of EA1045 and EAI001 was >50 µM and 14.5 ± 0.5 µM, respectively [9]. The allosteric ligand of EA1045 showed higher potency than EAI001. Both the allosteric ligands were used to identify the best homology model developed with the highest stability. The ligands were superimposed to identify the RMSD, which indicates the deviation in both ligands. The RMSD of the superimposed ligand was found to be 0.6Å, which means that there is less deviation between ligands of interest and can be used for docking studies.
EGFR homology model analysis
The ligand interaction study showed that the entire homology model developed had hydrogen bond interaction and π−π stack interaction with the amino acid of the selected EGFR protein. This interaction shows that the crystal structure developed was stable and complete, as the water molecule did not enter the ligand-binding sites. The GlideScore of both the high-affinity molecules was docked, and the GlideScore was obtained. The GlideScore for all 10 homology models of 5ZWJ showed a better score than 5D41. Overall, homology models 1 (for protein 5ZWJ) and 10 showed the best results and were shortlisted for further studies. The GlideScore is shown in the table insert of Figure 2.
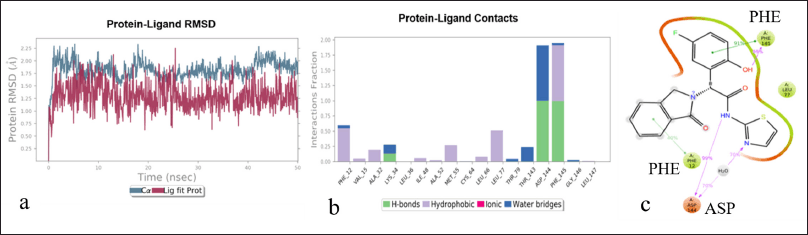 | Figure 2. Protein-ligand interaction in a simulated system a: protein-ligand interaction diagram at 50 ns. Pink lines represent ligand vibration, and blue lines represent protein vibration. b: The nature of the protein-ligand complex; three strong hydrogen bonds were observed. c: The protein-ligand interaction showing π-π amino acid PHE and two hydrogen bond formations at ASP and PHE. [Click here to view] |
The model was also analyzed for its interaction studies between ligand and hydrogen bond; along with it, π−π stack interaction was also observed in the homology model. Therefore, homology model-10 was finalized as our crystal structure for analysis for docking studies. This confirms that these homology models can be used for docking analysis (Table 1).
The Y-axis of the RMSD plot shows the stability of the ligand with the protein complex. The graph obtained in Figure 3 is “Lig-Fit-Prot,” showing the interaction complex between the ligand and protein. The protein−ligand graph is desired to show a slight deviation to avoid undesirable binding defects and to achieve strong interactions (Fig. 2a). The protein−ligand interaction result states that the RMSD plot of the protein−ligand is 1.3Å. However, after 30 ns, the RMSD value of the protein−ligand was found to be 0.75 Å. Therefore, the ligand has a stable interaction with the protein. The simulation analysis method was used to study the protein interactions with the ligand. This simulation analysis helps identify and quantify the hydrophobic and hydrogen bonds and the ionic and water bridge interactions. The values with a bar less than or equal to 0.7 indicate that 70% of the time, simulation interactions were maintained in the protein−ligand complex. The values above 0.7 bar value show that some part of the ligand interacts with multiple amino acids in the protein complex (Fig. 2b). The 2D analysis of the protein−ligand complex showed a strong affinity with the allosteric site. These interactions have stability, and two π−π bonds strongly interact with the amino acid sequence. Ligand atom interaction analysis is shown in Figure 2c. The protein−ligand contact with more than 30% of the simulation timeline is desired and likely to show potential allosteric inhibitory activity at the EGFR protein. The homology model was finalized for the visual screening method based on the abovementioned results. Additionally, the model was finalized for visual screening analysis of FDA molecules using consensual and normal docking.
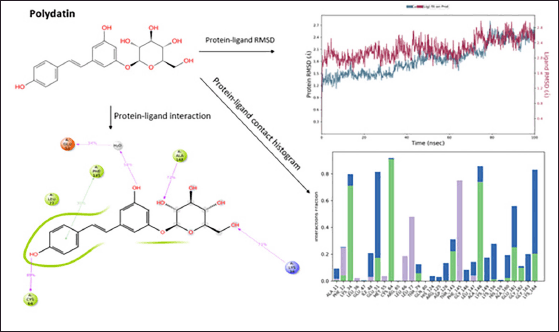 | Figure 3. Molecular Dynamics simulation of polydatin. [Click here to view] |
Virtual screening analysis by consensual docking
The most frequently repeating molecules within the first 20 XP results were identified, and their chemical properties were analyzed. The best molecules frequently repeating in all five frames, namely A, B, C, D, and E, are identified and listed in Table 2. The model that did not produce any results was excluded from further study.
 | Table 2. Consensual docking data with the models’ best compounds.* [Click here to view] |
MD simulation study: molecules shortlisted by consensual docking
Molecular docking is a computer-aided drug design technique that provides accurate and specific insights into structural biology when it interacts with a drug molecule. This analysis is important as it facilitates the input about the optimal drug−receptor protein interaction, which is the major focus of this study. Various other critical information like RMSD data is obtained via MD simulation study, which provides valuable insights about the residual flexibility of the protein’s backbone. This is essential for obtaining stability confirmation of the protein−ligand complex [26]. Therefore, MD simulation analysis is crucial for all the in silico studies, to understand the action of the test or target drugs on the biological systems. In this study, MD study was performed of the target molecules to understand its interaction pattern with the allosteric site of EGFR TK in case of NSCLC.
MD simulation analysis of Polydatin
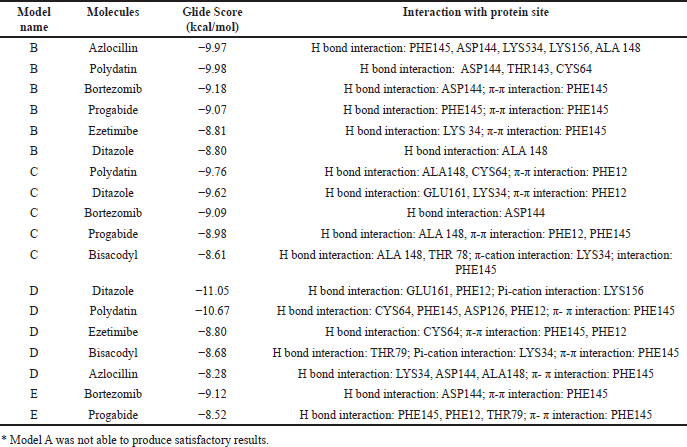
Polydatin showed a good protein−ligand interaction as it had H-bonds with amino acids of the allosteric sites ALA148, LYS34, CYS64, and GLU51 during the molecular dynamic simulation for 100 ns. A π−π interaction was observed with the amino acid PHE145. The compound shows good hydrophobicity throughout 100 nanoseconds (ns) ligand−protein interaction, as shown in the histogram in blue. The RMSD range of ligand−protein interaction was observed to be between 2.7 and 2.8 Å. The RMSD interactions were observed at 15–20, 30–40, and 50–100 ns. Figure 3 represents the pattern of the protein−ligand interaction during molecular dynamic simulations.
MD simulation analysis of Ezetimibe polydatin
Ezetimibe was the second molecule that showed good protein−ligand interaction, forming H-bonds with LEU127, TRP169, LYS 168, and ARG130. The π−cation interaction was observed with the amino acid LYS169. The ezetimibe shows good hydrophobicity throughout 100 ns ligand−protein interaction, as shown in the histogram in blue. The RMSD range of ligand−protein interaction was between 2.2Å and 2.8 Å. The RMSD interactions were observed throughout 100 ns. The compound shows a good interaction pattern with the protein (Fig. 4).
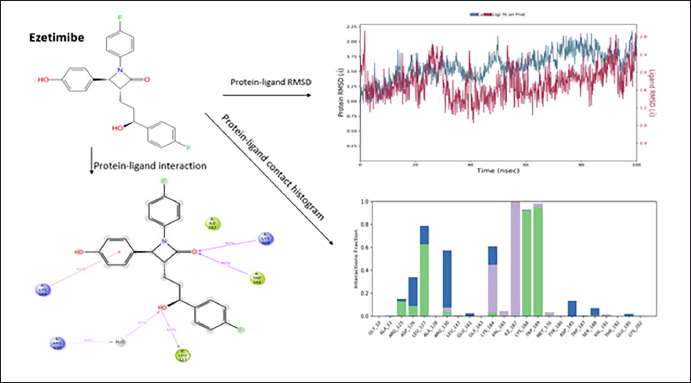 | Figure 4. Molecular Dynamics simulation of ezetimibe. [Click here to view] |
The compound with the anticipated interaction with the allosteric site EGFR protein was identified. The glide score, dock score, and interaction pattern were analyzed (Fig. 5). Table 3 shows the properties of the top compounds when in interaction with the protein. Based on the protein−ligand interaction score and interaction analysis, the top compounds were identified and then analyzed in MD simulation studies to identify the best compound (Table 3).
 | Figure 5. Homology model-10 with EGFR allosteric site for normal virtual screening analysis. [Click here to view] |
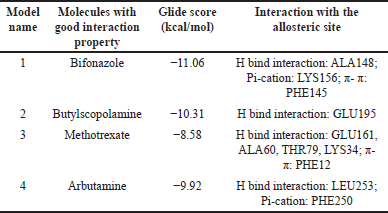 | Table 3. Virtual screening results of the FDA-approved molecules using the normal docking method on homology Model-10. [Click here to view] |
MD simulation study: molecules shortlisted by normal docking
MD simulation analysis of methotrexate
Methotrexate shows fair protein−ligand interaction forming H-bonds with ALA148; Pi−cation interaction is observed at site LYS156, and π−π interaction is observed at site PHE12. The compound shows good hydrophobicity ligand−protein interaction, as shown in the histogram in blue. The RMSD range of ligand−protein interaction was between 2.8 and 4.0 Å. The RMSD interactions were observed throughout 0–100 ns. The compound produced satisfactory results and will be further investigated for pharmacological evaluation (Fig. 6).
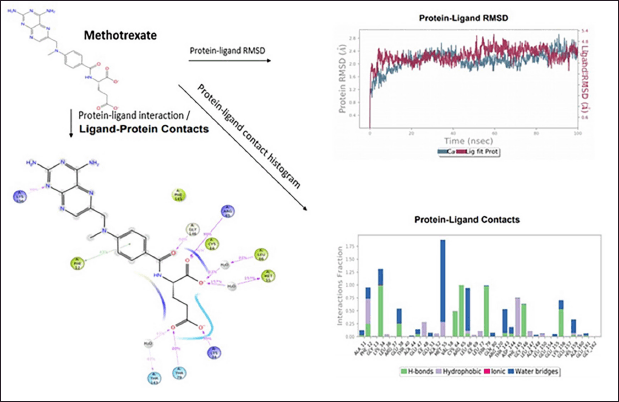 | Figure 6. Molecular Dynamics simulation of methotrexate. [Click here to view] |
MD simulation analysis of arbutamine
The drug arbutamine shows fair protein−ligand interaction forming H-bonds with ASP185, GLU118, HIS182, and PRO248, and π−π interaction is observed at site PHE250. The compound shows good hydrophobicity ligand−protein interaction, as shown in the histogram in blue. The RMSD range of ligand−protein interaction was between 2.4 and 4.5 Å. The RMSD interactions were observed throughout 0–100 ns. The compound produced satisfactory results and will be further investigated for pharmacological evaluation (Fig. 7).
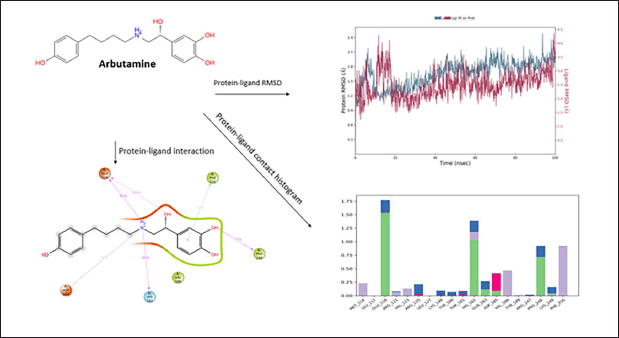 | Figure 7. Molecular Dynamics simulation of Arbutamine. [Click here to view] |
DISCUSSION
In this pandemic, due to the coronavirus (COVID-19) outbreak, NSCLC patient’s mortality rate has been reported to have drastically increased [27]. To solve this problem, researchers must use new methodologies to develop new therapies quickly and curb cost-effectiveness [28]. We have conducted a similar study of drug repurposing utilizing tools in Schrodinger software for predicting anti-COVID-19 drugs [25,28]. In NSCLC, the available therapy targets RTKs, which is challenging and often risky due to readily acquired resistance development in the patients [27]. This condition may also lead to mutation in the amino acid sequence of the RTKs. It is reported that the first-generation EGFR inhibitors lead to overexpression of CYP3A4, leading to hepatotoxicity and multiple liver dysfunctions [29]. The consumption of second-generation EGFR inhibitors leads to T790M mutation, causing drug resistance in 60% of the patients [30]. Even though third-generation drugs overcome the T790M mutation, stopping the therapy might lead to developing the C797s mutation condition in most patients [31]. Therefore, the paramount must do in this condition is identifying fresh indications with the least risk of drug resistance development due to instability in EGFR RTKs complex leading to T790M/C797S mutations [32]. Allosteric sites of EGFR inhibitors like EAI045 can potentially terminate the aggressive growth of NSCLC harboring EGFR L858R/ T790M/ C797s. In addition, allosteric inhibitors are likely to cause lower toxicity and provide more stable biological interaction in conjugate with cetuximab [33]. Therefore, biochemical optimization analysis of the EGFR allosteric region can be a promising therapeutic outcome for inhibiting the fatal NSCLC growth for patients in the advanced stages.
Two new allosteric inhibitors of EGFR, namely EAI001 and EAI045, were reported as successful targets to achieve the drug resistance and mutation conditions in NSCLC cases. We designed a study to identify independent EGFR allosteric inhibitors that can overcome the shortcomings of the available therapies. Drug re-purposing is proven to be an efficient technique for the identification of pharmacotherapies by investing little capital. Molecular docking, followed by MD simulation analysis, are widely accepted techniques to obtain information about the drug molecule−ligand interaction during the drug discovery process [34]. Molecular docking of 2,800 drugs was carried out on the homology protein developed. We carried out a two-way docking study method: (i) consensual docking study, where the five most stable poses of the obtained homology complex were identified and used for MD analysis and (ii) normal docking, where the FDA molecules were docked on the developed homology model. After the protein preparation method, the homology model was subjected to docking analysis. The ligands were docked using the extra precision method. Homology model-10 showed better results and a gradual decrease in glide score. Further confirmation of the homology models was achieved by performing an MD simulation study. These docking methods gave us a better idea about the repurposed compounds and increased the chances of obtaining more potent anti-NSCLC compounds. The virtual screening analysis, docking results, and MD simulation studies on the FDA-approved molecules were conducted and optimized. Ezetimibe, polydatin, methotrexate, and arbutamine were identified to be potent as EGFR allosteric site inhibitors. Ezetimibe is the first known selective cholesterol-absorption inhibitor that stops the cholesterol uptake from the intestinal lumen with negligible effect on other sterols or lipid-soluble vitamins [35,36]. In another study, ezetimibe showed hydrogen bond interactions with the allosteric site of EGFR at LEU127, TRP169, LYS168, and ARG130, which is required for stable interaction with the target site. These inhibitions will ultimately lead to controlled auto-phosphorylation in the RTKs. This will stop the mutation development condition [9].
Polydatin, also chemically stated as, 3,4,5-trihydroxystilbene-3-β-D-glucoside, is an active constituent from natural products such as grapes, peanuts, and cocoa-containing products with multiple pharmacological effects such as anti-inflammatory, immunoregulatory, lung protective and antioxidant effects [37,38]. The drugs cetuximab and panitumumab, which are therapeutically used extracellular EGFR inhibitors, and drugs such as gefitinib, erlotinib, and afatinib, which are intracellular EGFR inhibitors, produce dermatological reactions called papulopustular eruption. A recent clinical study reported that skin adverse reactions can be treated by the topical application of polydatin 0.8% and 1.5% polydatin cream [39]. These properties make it one of the most suitable compounds to target EGFR allosteric sites in NSCLC. Our study shows that polydatin has H-bond interactions with ALA148, LYS34, CYS64, and GLU51 at the allosteric site. π−π interaction was observed with amino acid PHE145 of the allosteric site. The compound shows good hydrophobicity throughout the 100 ns ligand−protein interaction. These results are supported by its good anti-inflammatory and immunological activity in the lungs [38]. Therefore, it can be a good candidate drug for developing an EGFR allosteric inhibitor.
Methotrexate is widely known as an anti-arthritis agent with complex action. However, the well-known hypothesis for its pharmacological activity is the anti-inflammatory effect due to adenosine release. It also increases the T cells and cytokine, leading to apoptosis, and it is therapeutically used to treat NSCLC in combination [40]. However, because of better drugs currently, methotrexate is not preferred in NSCLC. We prove here that the properties of alteration of the cell signaling pathway can be the reason for interaction with the allosteric site of EGFR in NSCLC conditions. In our study, methotrexate shows strong H-bonds with ALA148; Pi−cation interaction is observed at site LYS156, and π−π interaction is observed at site PHE12. These interactions are desired for EGFR allosteric inhibition [10]. Therefore, it can serve as a potent EGFR allosteric inhibition agent and act as an anti-NSCLC agent.
Arbutamine is a popular cardiac agent classified as a βeta-adrenergic blocker, specially developed for cardiac stress testing [41]. Arbutamine showed EGFR allosteric inhibitory effect in the current in silico docking and MD simulation studies. The study showed that arbutamine has protein−ligand interaction, forming H-bonds with ASP185, GLU118, HIS182, and PRO248, and a π−π interaction is observed at site PHE250. The compound shows good hydrophobicity in ligand−protein interactions. This property makes it suitable as a potent allosteric agent to inhibit EGFR in NSCLC conditions. All the hits obtained in the study have not been reported for any activity against NSCLC.
Chemotherapy has been the popular choice for NSCLC remedy, and therefore, the identified EGFR allosteric compounds can be further explored for various combination therapies with cisplatin or platinum-based compounds such as carboplatin [42] or the evaluation of these drugs to injectable platinum-based therapy forms can be explored [43]. Therefore, these drugs can be further explored for their in vitro and in vivo study to identify the best two molecules that can serve as a fourth-generation EGFR allosteric inhibitor in treating NSCLC.
CONCLUSION
The present study successfully identified and validated potential allosteric inhibitors for the EGFR protein through an integrated computational approach involving homology modeling, molecular docking, and MD simulations. Among the prepared ligands, EA1045 demonstrated superior allosteric potency and structural stability, supported by its low RMSD value (0.6 Å) and GlideScores in docking studies. Homology model-10 was finalized due to its stable hydrogen bonding and π−π interactions, providing a reliable crystal structure for virtual screening. Through consensual and normal docking, polydatin and ezetimibe emerged as top candidates, showing consistent and strong protein−ligand interactions over 100 ns simulations. Methotrexate and arbutamine, identified via normal docking, also showed promising yet comparatively moderate stability. Overall, the computational findings indicate that polydatin and ezetimibe are strong candidates for further in vitro and in vivo validation as allosteric inhibitors of EGFR, potentially contributing to novel therapeutic strategies in EGFR-targeted therapies. The newly designed in silico methodology for drug screening/repurposing to identify new indications for the target disease showed that the drugs polydatin, ezetimibe, methotrexate, and arbutamine could be potent EGFR allosteric site inhibitors.
The findings drew the way to find out new pharmacotherapies from the available drugs at a faster pace compared to traditional drug discovery methods. The utilization of in silico approach to tackle the fatal NSCLC can be a meaningful research advancement. However, further in vitro and in vivo investigations of these hits could lead to identifying the most potent allosteric EGFR inhibitors for NSCLC.
ACKNOWLEDGMENT
The authors are grateful to the ICMR, New Delhi, for providing a Senior Research Fellowship (ICMR-SRF #45/33/2019-PHA/BMS) to Swastika Maity. The authors also thank the Department of Pharmaceutics, Manipal College of Pharmaceutical Sciences, for facilitating the computer simulations. Schrodinger’s Software and Computers were procured under the grant from DST-SERB, New Delhi, to Dr Usha Y Nayak (EMR/2016/007006).
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to the conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be author as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included in this research article.
PUBLISHER’S NOTE
All claims expressed in this article are solely those of the authors and do not necessarily represent those of the publisher, the editors and the reviewers. This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
USE OF ARTIFICIAL INTELLIGENCE (AI)-ASSISTED TECHNOLOGY
The authors declares that they have not used artificial intelligence (AI)-tools for writing and editing of the manuscript, and no images were manipulated using AI.
REFERENCES
1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin. 2021;71:7–33. CrossRef
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30. CrossRef
3. Carcereny E, Morán T, Capdevila L, Cros S, Vilà L, de los Llanos Gil M, et al. The epidermal growth factor receptor (EGRF) in lung cancer. Transl Respir Med. 2015;3:1–8. CrossRef
4. Sullivan I, Planchard D. Next-generation EGFR tyrosine kinase inhibitors for treating EGFR-mutant lung cancer beyond first line. Front Med. 2016;3:1–13. CrossRef
5. Roskoski RJr. Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol Res. 2016;103:26–48. CrossRef
6. Maity S, Pai KSR, Nayak Y. Advances in targeting EGFR allosteric site as anti-NSCLC therapy to overcome the drug resistance. Pharmacol Rep. 2020;72:799–813. CrossRef
7. Purba E, Saita E, Maruyama I. Activation of the EGF Receptor by ligand binding and oncogenic mutations: the “Rotation Model.” Cells. 2017;6:13. CrossRef
8. Ling Y, Jing M, Wang X dong. Allosteric therapies for lung cancer. Cancer Metastasis Rev. 2015;34:303–12. CrossRef
9. Jia Y, Yun CH, Park E, Ercan D, Manuia M, Juarez J, et al. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature. 2016;534:129–32. CrossRef
10. Zhao P, Yao MY, Zhu SJ, Chen JY, Yun CH. Crystal structure of EGFR T790M/C797S/V948R in complex with EAI045. Biochem Biophys Res Commun. 2018;502:332–7. CrossRef
11. Wang S, Song Y, Liu D. EAI045: the fourth-generation EGFR inhibitor overcoming T790M and C797S resistance. Cancer Lett. 2017;385:51–4. CrossRef
12. Caporuscio F, Tinivella A, Restelli V, Semrau MS, Pinzi L, Storici P, et al. Identification of small-molecule EGFR allosteric inhibitors by high-Throughput docking. Fut Med Chem. 2018;10:1545–53. CrossRef
13. Saipriya DS, Prakash A, Kini SG, Bhatt GV, Ranganath Pai KS, Biswas S, et al. Design, synthesis, antioxidant and anticancer activity of novel schiff’s bases of 2-amino benzothiazole. Indian J Pharm Educ Res. 2018;52:S333–42. CrossRef
14. Chen IJ, Foloppe N. Drug-like bioactive structures and conformational coverage with the ligprep/confgen suite: comparison to programs MOE and catalyst. J Chem Inf Model. 2010;50:822–39. CrossRef
15. Roos K, Wu C, Damm W, Reboul M, Stevenson JM, Lu C, et al. OPLS3e: Extending force field coverage for drug-like small molecules. J Chem Theory Comput. 2019;15:1863–74. CrossRef
16. Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Computer-Aided Mol Design. 2013;27:221–34. CrossRef
17. Rostkowski M, Olsson MH, Søndergaard CR, Jensen JH. Graphical analysis of pH-dependent properties of proteins predicted using PROPKA. BMC Structural Biol. 2011;11:6. CrossRef
18. Subhani S, Jayaraman A, Jamil K. Homology modelling and molecular docking of MDR1 with chemotherapeutic agents in non-small cell lung cancer. Biomed Pharmacother. 2015;71:37–45. CrossRef
19. Jacobson MP, Pincus DL, Rapp CS, Day TJF, Honig B, Shaw DE, et al. A hierarchical approach to all-atom protein loop prediction. Proteins: Struct Funct Bioinf. 2004;55:351–67. CrossRef
20. Ramachandran GN, Ramakrishnan C, Sasisekharan V. Stereochemistry of polypeptide chain configurations. J Mol Biol. 1963;7:95–9. CrossRef
21. Isa AS, Uzairu A, Umar UM, Ibrahim MT, Umar AB, Tabti K, et al. In silico exploration of novel EGFR-targeting compounds: integrative molecular modeling, docking, pharmacokinetics, and MD simulations for advancing anti-cervical cancer therapeutics. Sci Rep. 2025;15:7334. CrossRef
22. Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, et al. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem. 2004;47:1750–9. CrossRef
23. Damm-Ganamet KL, Arora N, Becart S, Edwards JP, Lebsack AD, McAllister HM, et al. Accelerating lead identification by high throughput virtual screening: prospective case studies from the pharmaceutical industry. J Chem Inf Model. 2019;59:2046–62. CrossRef
24. Akash S, Islam MR, Bhuiyan AA, Islam MN, Bay?l I, Saleem RM, et al. In silico evaluation of anti-colorectal cancer inhibitors by Resveratrol derivatives targeting Armadillo repeats domain of APC: molecular docking and molecular dynamics simulation. Front Oncol. 2024;14:1360745.
25. Baby K, Maity S, Mehta CH, Suresh A, Nayak UY, Nayak Y. Targeting SARS-CoV-2 RNA-dependent RNA polymerase: an in silico drug repurposing for COVID-19. F1000Res. 2020;9:1166. CrossRef
26. Yu H, Dalby PA. Coupled molecular dynamics mediate long-and short-range epistasis between mutations that affect stability and aggregation kinetics. Proc Natl Acad Sci. 2018;115:E11043–52. doi: 10.1073/pnas.1810324115
27. Passaro A, Bestvina C, Velez Velez M, Garassino MC, Garon E, Peters S. Severity of COVID-19 in patients with lung cancer: evidence and challenges. J Immunother Cancer. 2021;9(3):e002266. CrossRef
28. Rogado J, Pangua C, Serrano-Montero G, Obispo B, Marino AM, Pérez-Pérez M, et al. Covid-19 and lung cancer: a greater fatality rate? Lung Cancer. 2020;146:19–22. CrossRef
29. Baby K, Maity S, Mehta CH, Suresh A, Nayak UY, Nayak Y. SARS-CoV-2 entry inhibitors by dual targeting TMPRSS2 and ACE2: an in silico drug repurposing study. Eur J Pharmacol. 2021;896:173922. CrossRef
30. Sutto L, Gervasio FL. Effects of oncogenic mutations on the conformational free-energy landscape of EGFR kinase. Proc Natl Acad Sci U S A 2013;110:10616–21. CrossRef
31. Kim MK, Yee J, Cho YS, Jang HW, Han JM, Gwak HS. Risk factors for erlotinib-induced hepatotoxicity: a retrospective follow-up study. BMC Cancer. 2018;18:1–7. CrossRef
32. Zhang Y, Wang C, Liu Z, Meng Q, Huo X, Liu Q, et al. P-gp is involved in the intestinal absorption and biliary excretion of afatinib in vitro and in rats. Pharmacol Rep. 2018;70:243–50. CrossRef
33. Fassunke J, Müller F, Keul M, Michels S, Dammert MA, Schmitt A, et al. Overcoming EGFR G724S -mediated osimertinib resistance through unique binding characteristics of second-generation EGFR inhibitors. Nat Commun. 2018;9:4655. CrossRef
34. Baby K, Maity S, Mehta CH, Nayak UY, Shenoy GG, Pai KSR, et al. Computational drug repurposing of Akt-1 allosteric inhibitors for non-small cell lung cancer. Sci Rep. 2023;13(1):7947. CrossRef
35. Leonetti A, Sharma S, Minari R, Perego P, Giovannetti E, Tiseo M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br J Cancer. 2019;121:725–37. CrossRef
36. Gupta EK, Ito MK. Ezetimibe: The first in a novel class of selective cholesterol-absorption inhibitors. Heart Dis. 2002;4:399–409. CrossRef
37. Balzer BWR, Loo C, Lewis CR, Trahair TN, Anazodo AC. Adenocarcinoma of the lung in childhood and adolescence: a systematic review. J Thoracic Oncol. 2018;13:1832–41. CrossRef
38. Du QH, Peng C, Zhang H. Polydatin: a review of pharmacology and pharmacokinetics. Pharm Biol. 2013;51:1347–54. CrossRef
39. Bavetta M, Silvaggio D, Campione E, Sollena P, Formica V, Coletta D, et al. The effects of association of topical polydatin improves the preemptive systemic treatment on EGFR inhibitors cutaneous adverse reactions. J Clin Med. 2021;10:1–8. CrossRef
40. Smyth JF, Ford HT. Methotrexate in the chemotherapy of lung cancer. Cancer Treatment Rep. 1981;65:161–3.
41. Ketteler T, Krahwinkel W, Wolfertz J, Godke J, Hoffmeister T, Scheuble L, et al. Arbutamine stress echocardiography. Eur Heart J. 1997;18:D24–30. CrossRef
42. Khokhar FM, Jahangir TM, Khuhawar MY, Khaskheli MI, Khokhar LA, Abro MI, et al. Analysis of platinum-based anticancer injections cisplatin and carboplatin in blood serum and urine of cancer patients by photometry, fluorometry, liquid chromatography using a Schiff-base as derivatizing reagent. J Pharm Biomed Anal. 2024;238:115808. Available from: https://www.sciencedirect.com/science/article/pii/S0731708523005770
43. Khokhar FM, Jahangir TM, Khuhawar MY, Qureshi MS, Khaskheli MI, Khokhar LAK. High performance liquid chromatographic separation of platinum (II), gold (III), vanadium (IV), vanadium (V), molybdenum (VI) and analysis of cis-platin as platinum (II) in cis-plasol injection, urine, and blood serum using pyridoxal-4-phenyl-3-thiosemicarbazone as complexing reagent. J Liq Chromatogr Relat Technol. 2020;43(1–2):29–36. CrossRef