
INTRODUCTION
The nonsteroidal anti-inflammatory drug indomethacin is used to treat known diseases or for medical procedures. Rheumatoid arthritis is a systemic autoimmune disease characterized by chronic joint inflammation. The incidence among women is 2–3 times higher than that in men. Traditional drug therapy for this disease has many disadvantages like poor bioavailability and degradation by gastrointestinal enzymes [1]. On the other hand, endoscopic retrograde cholangiopancreatography (ERCP) is a widely used medical procedure for the diagnosis of liver, bile duct, and pancreatic problems. Indomethacin is a drug of choice for the prevention of post-ERCP pancreatitis [2,3].
According to the biopharmaceutical classification system, indomethacin belongs to Class II drugs [4]; hence, it exhibits a fast absorption rate but lacks good solubility. The mechanism of action of indomethacin is well documented, and it is known that indomethacin acts as a non-selective cyclooxygenase (COX) inhibitor. Inhibition occurs in COX-1 and COX-2 isoforms. The COX-2 isoform is activated at inflammation sites, whereas the COX-1 isoform is constitutive and regulates normal body functions [5].
Rectal administration is recommended for patients who are unconscious, hesitate, vomit, or difficult to swallow. This route of administration avoids the first-pass effect or presystemic metabolism. However, low/variable absorption due to high interindividual variability has been documented [6]. An absolute bioavailability factor f = 0.8 was reported for rectal indomethacin administration [7].
To ensure acceptable biopharmaceutical quality in the post-marketing evaluation, dissolution studies are an adequate option to reveal differences in the release rate, which may prevent therapeutic failure. Some authors have reported the preparation of rectal formulations and their evaluation with in vitro release studies to improve drug absorption and therapeutic effects [8,9]. Indomethacin suppositories are commercially available as over-the-counter drugs.
The indomethacin suppository pharmacopeial dissolution test established a USP paddle apparatus (USP apparatus II) at an agitation rate of 50 rpm and 900 ml of phosphate buffer (0.1 M, pH 7.2). At 60 minutes, not less than 75% of the labeled amount is dissolved [10]. In addition, the mathematical management of dissolution data are a valuable tool for simulating plasma concentration-time profiles. Convolution is a well-known model-independent approach that helps simulate human performance. The hypothetical values of Cmax and AUC0–inf were calculated using in vitro data (as input functions) as well as pharmacokinetic parameter values of innovator drug products (as a weighted function) [11].
To date, no hypothetical plasma levels of indomethacin suppositories from dissolution data generated by the USP basket apparatus and flow-through cell method (USP apparatus IV) have been reported. The USP apparatus IV is a special device for testing rectal formulations manufactured with poorly water-soluble drugs. Some studies have shown good in vitro/in vivo correlation with dissolution data generated by the flow-through cell method [12,13]. Therefore, it is important to document the usefulness of this USP apparatus for simulating indomethacin in vivo performance using semisolid formulations.
The objective of this study was to predict the indomethacin plasma levels of all available suppositories from the local market. Hypothetical in vivo performance was calculated using the percentage of indomethacin dissolved obtained using USP apparatuses I and IV as well as pharmacokinetic parameters (elimination rate constant, volume of distribution, and bioavailability factor f) using a numerical convolution method. Results could be important in proposing better semisolid rectal formulations manufactured with indomethacin.
MATERIAL AND METHODS
Chemicals
Only two indomethacin semisolid rectal dosage forms (suppositories, 100 mg) available in the local market were used. The reference (classified as R drug product) and generic formulation (classified as G formulation) were tested. J.T.Baker High Performance Liquid Chromatography methanol (≥99.9%), AR sodium hydroxide (≥98%), AR sodium phosphate monobasic crystals (98%–102%), and sodium dodecyl sulfate (SDS) (≥99%) were acquired from a local supplier (Mexico). The indomethacin reference compound was acquired from Sigma-Aldrich Co. (purity 98.5%–100.5%, St. Louis, MO). In each experiment, five indomethacin solutions of known concentration (3.12–50 µg/ml) in phosphate buffer (0.1 M, pH 7.4) were prepared.
USP basket apparatus
The dissolution profiles of indomethacin suppositories were obtained using USP apparatus I (Sotax AT-7 Smart Model, Switzerland) at an agitation rate of 100 rpm. To quantify released indomethacin, spectrophotometric analysis was considered (Perkin Elmer spectrophotometer Lambda 35 Model, USA). The dissolved drug was calculated using a calibration curve with indomethacin standard solutions and UV absorbance determination at 318 nm. The formulations were added to 750 ml of phosphate buffer (0.1 M, pH 7.4). To improve in vitro release, the same medium was added with 1% SDS. The temperature of the dissolution medium was 37.0°C ± 0.5°C. The percentage of dissolved indomethacin was calculated every 5 over 60 minutes (n = 12) with the support of standard solutions (with or without 1% SDS).
Flow-through cell method
Indomethacin suppositories were tested using United States Pharmacopeia (USP) apparatus IV with specific dual chamber suppository cells (Sotax CE6 Model, Switzerland). As dissolution medium, phosphate buffer (0.1 M, pH 7.4) or phosphate buffer (0.1 M, pH 7.4) was added with 1% SDS and pumped at 16 ml/min. Dissolved indomethacin was calculated spectrophotometrically at the same sampling times as in the USP apparatus I experiments.
Data treatment
The dissolution profiles of indomethacin in the formulations were compared with model-independent and model-dependent methods. According to Equation 1, similarity factor f2 was calculated. An f2 value between 50–100 has been established to ensure the sameness of the two in vitro release curves [14]. In addition, the dissolution efficiency (DE), dissolved indomethacin level at 60 minutes (Q60), and mean dissolution time (MDT) were determined. The f2, MDT, and DE values were computed with the support of the add-in DDSolver [15]. Results were analyzed using Student’s t-test. The mechanism of indomethacin release from suppositories was determined by fitting dissolution data with several mathematical equations. The following models were used: First-order, Makoid–Banakar, Korsmeyer–Peppas, Peppas–Sahlin, and Weibull. Fittings were computed using the add-in DDSolver [15]. To mathematically explain the in vitro release mechanism, the model that showed the highest adjusted determination coefficient (R2adjusted) and the lowest akaike information criterion (AIC) was selected as the best-fit model [16].
(1)
Here, n is the number of time points, and Rt and Tt are the dissolution values of the reference and test product at time t, respectively [14].
Hypothetical in vivo behavior
Dissolution profiles generated by both USP apparatuses and published pharmacokinetic information [7] were used to predict indomethacin in vivo behavior. A simple numerical convolution method was used [17]. The predicted plasma concentrations were fitted with a two-compartment open model using the add-in PKSolver [18]. Peak plasma concentration (Cmax) and area under the curve from zero time to infinity (AUC0–inf) were computed and related to observed pharmacokinetic parameters. The observed Cmax (2.36 µg/ml) and AUC0–inf (9.08 µgh/ml) were calculated after adjusting indomethacin in vivo data [7]. The predictability of the convolution method was tested using the percentage of prediction error (%PE) for Cmax and AUC0–inf. The %PE was calculated using Equation 2 (suitable values should not exceed 10%) [19]. All calculations were performed using an Excel spreadsheet.
(2)
RESULTS AND DISCUSSION
Standard calibration curve
Indomethacin standard calibration curves in phosphate buffer (0.1 M, pH 7.4) with and without the addition of 1% SDS were linear in the range of 3.12–50 µg/ml. The linear regression results are shown in Figure 1. Both linear regressions were significant (p < 0.05).
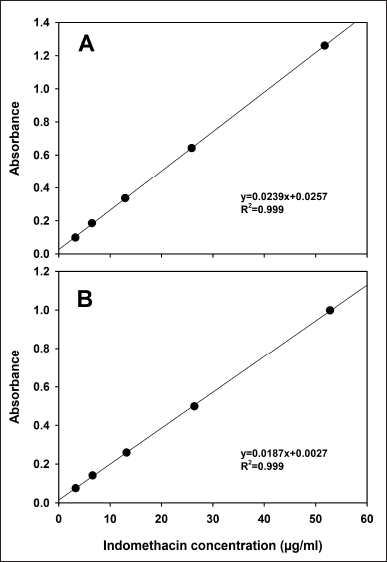 | Figure 1. Standard calibration curves in phosphate buffer (0.1 M, pH 7.4) (A) without 1% SDS and (B) added with 1% SDS. Mean, n = 3. [Click here to view] |
In vitro release data
The indomethacin dissolution profiles of the R and G formulations are depicted in Figure 2. Both drug products exhibited different in vitro release performances. The release pattern of the R drug product remained consistent, regardless of the dissolution conditions. The G formulation showed high sensitivity to the hydrodynamic environment of both dissolution apparatuses, and a faster in vitro release was observed using the flow-through cell method. The addition of a surfactant did not appear to modify the release of indomethacin. The f2 values are given in Table 1. In all cases, no similarity in dissolution profiles between the G and R formulations was observed (f2 < 50).
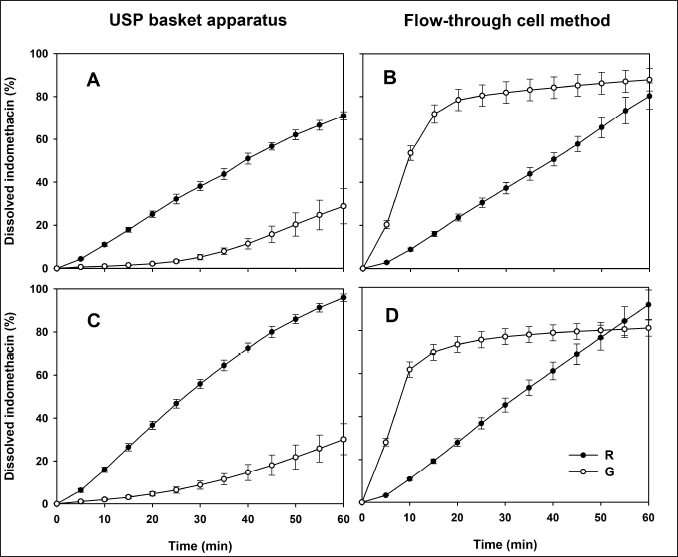 | Figure 2. Dissolution profiles of indomethacin suppositories with phosphate buffer (0.1 M, pH 7.4). (A and B) Profiles where no 1% SDS was added. (C and D) Profiles with 1% SDS in dissolution medium. Reference (R) and generic (G) formulation. Mean ± SD, n = 12. [Click here to view] |
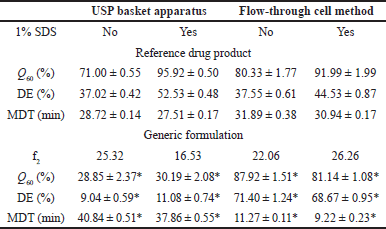 | Table 1. Similarity factor f2 and dissolution parameters. Mean ± SE medium, n = 12. *p < 0.05. [Click here to view] |
Considering the use of surfactants, some authors have shown that three mechanisms are involved in the increase of drug release: improved wetting, solubilization, and the dissolution of soluble surfactants to form pores in the matrix [20]. On the other hand, several authors have found that indomethacin dissolution is faster in hydrophilic than in lipophilic suppositories. The distinction was more evident when the flow-through cell method was used [21]. Other authors stated that conventional USP apparatus I, paddle (II), or IV seem to be adequate for testing hydrophilic suppositories, while modified basket, modified paddle, or modified flow-through cell have been recommended for use with lipophilic suppositories [22]. In this study, only two commercially available pharmaceutical drug products were used, and information on the nature of the excipients or the manufacturing process is unknown. Both drug products showed a total difference in the in vitro release rate of the active ingredient, which suggests that one formulation is prepared with a hydrophilic base while the other with a lipophilic base. This assumption allows manufacturers to evaluate the impact of the nature of the drug based on indomethacin release and its repercussions on in vivo performance.
Results of dissolved indomethacin at the last sampling time (Q60), MDT data, and DE values are presented in Table 1. All Q60, DE, and MDT values of the G formulation differed significantly from the parameters of the R drug product in both USP dissolution apparatuses (p < 0.05). Considering the in vitro release behavior shown in Figure 2, these results were expected. Dissolution profile comparison using MDT data is a common approach and has been previously considered [23].
Our results are comparable with those of a study on sustained release of indomethacin suppositories. Experiments were carried out with USP apparatus I at 100 rpm and 900 ml of phosphate buffer (0.2 M, pH 7.2) as the dissolution medium. Control conventional suppositories released 98.43% ± 0.01% at 30 minutes [9], whereas in our work, the R drug product in the same USP apparatus but with dissolution media added with 1% SDS achieved 95.92 % at 60 minutes.
A study in which a continuous flow-through bead bed dissolution apparatus and rotating paddle method were used to test indomethacin suppositories (100 mg) from fatty and water-soluble bases was reported by some authors [24]. Polyethylene glycol water-soluble bases gave faster drug release rates compared to fatty bases, whereas the drug release rate from fatty bases increased significantly when a surface-active agent was present. On the other hand, the authors stated that the rotating paddle technique may be considered a suitable method for routine dissolution testing, but the continuous flow-through bead bed dissolution apparatus is more suitable for the experimental study of suppositories as this equipment significantly prolongs the drug release rate from suppositories and with this method, a better correlation may be obtained with the in vivo absorption.
To document the mechanism of indomethacin release from suppositories, the dissolution data of both drug products were fitted with common mathematical models as previously described. Results of R2adjusted and AIC are presented in Table 2.
All data generated by the R drug product in both hydrodynamic environments (with and without 1% SDS) and G formulation in the flow-through cell (with and without 1% SDS) were adjusted to the Weibull function. Data for G formulation of USP apparatus I (with and without 1% SDS) adjusted to Makoid–Banakar model. Adjustment of the dissolution data to the Weibull function emphasizes the S-shape of the dissolution profile while the Makoid–Banakar model becomes identical to the Korsmeyer–Peppas model when parameter k is zero. It follows the sole diffusion mechanism. The “n” function governs the shape of the dissolution curve [25].
The flow-through cell has shown better discriminatory capacity than other dissolution apparatuses in determining the in vitro release rate of drugs with limited solubility [26], so it is necessary to evaluate dissolution conditions like those observed in the human gastrointestinal tract. Several authors have found a meaningful correlation with data obtained using USP apparatus IV [13,27], and it is important to consider the details that led them to this conclusion, such as the physicochemical information of each active ingredient as well as data mathematical handling. Training human resources in the collection of data obtained using the flow-through cell method and in the handling of the information generated by this equipment is necessary.
Simulation of plasma concentrations
To compare our results, data from an in vivo study [7] are presented in Figure 3. Indomethacin plasma concentrations were fitted to a two-compartment open model using the add-in PKSolver [18]. The observed Cmax and AUC0–inf values are described in the same plot. After applying a convolution approach with, in vitro release data from both USP apparatuses and published pharmacokinetic information, the hypothetical in vivo behavior of indomethacin suppositories is shown in Figure 4.
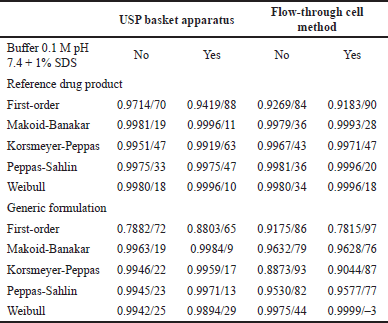 | Table 2. Values of R2adjusted/AIC. [Click here to view] |
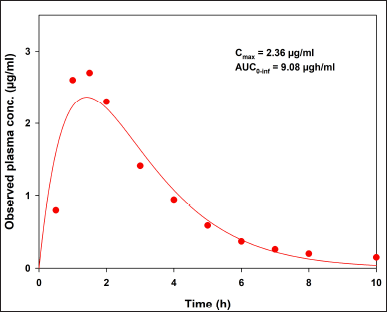 | Figure 3. Indomethacin plasma concentrations fitted to a two-compartment open model with the add-in PKSolver. Observed data previously reported [7] . [Click here to view] |
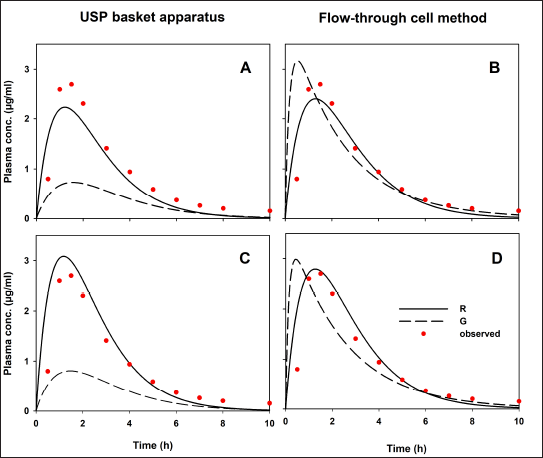 | Figure 4. Hypothetical indomethacin plasma concentration-time profiles. (A and B) Profiles where no 1% SDS was added. (C and D) Profiles with 1% SDS in dissolution medium. Reference (R) and generic (G) formulation. Observed data previously reported [7] . [Click here to view] |
Predicted human performances reflect the in vitro release behaviors generated by the hydrodynamic environment of both dissolution apparatus types. Values of predicted Cmax and AUC0–inf are shown in Table 3. As comparative data, some authors have studied the bioavailability of indomethacin suppositories (100 mg). Four formulations were administered to healthy volunteers, and drug levels were quantified. The reported Cmax and AUC0–12h for Indocid® formulation was 3.08 ± 0.96 (2.18–4.35) and 9.51 ± 4.06 (4.98–14.18) µgh/ml, respectively [28]. The PE values calculated to validate the convolution of dissolution data are presented in Table 3.
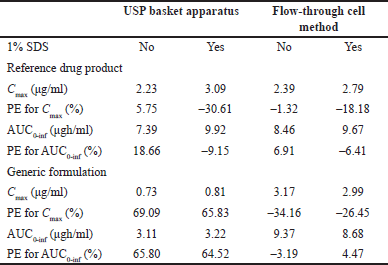 | Table 3. Hypothetical Cmax and AUC0-inf values and PE for each parameter calculated with indomethacin dissolution data and published pharmacokinetic information. [Click here to view] |
Only PE data <10% for the pharmacokinetic parameters Cmax and AUC0–inf of the R drug product was achieved using the flow-through cell method. Since this USP apparatus generates a hydrodynamic environment similar to that found in the gastrointestinal tract, the mathematical transformation of the dissolution data of R formulation adequately simulates drug absorption and generates Cmax and AUC0–inf values similar to an in vivo study (Fig. 3). If the R formulation of the reported human study has been manufactured without significantly changing the excipients or the manufacturing process, the PE values generated by the current in vitro data will be less than 10%. Appropriate conditions for evaluating the quality of indomethacin suppositories appear to be the use of USP apparatus IV at a flow rate of 16 ml/min and phosphate buffer (0.1 M, pH 7.4) without 1% SDS. To achieve comparable biopharmaceutical quality, multisource formulations should have dissolution profiles similar to those of R drug products under previously described in vitro conditions. In this regard, the concept of a bio-predictive dissolution tool can be used to estimate the human pharmacokinetic and support formulation design [29].
No hypothetical pharmacokinetic parameters calculated using dissolution data for the G formulation generated by the USP basket apparatus achieved PE values less than 10% (with or without 1% SDS). Therefore, in this case, USP apparatus I was not capable of generating in vitro release data that can be mathematically transformed into hypothetical concentrations like those reported in an in vivo study. These differences between the R and G formulations cannot be attributed to the convolution method, since it was applied in the same way to all data, nor to experimental variability, since the dissolution data comply with the variability allowed by international standards (coefficient of variation <20% at the earlier time points and <10% at other time points) [30]. The difference in the prediction of the drug’s performance in the G formulation, considering the PE values of both pharmacokinetic parameters, agrees with the evident difference in the rate and extent of drug release from the pharmaceutical dosage form that contains it.
Some advantages of using USP apparatus IV as an alternative method to the vessels apparatuses (USP I and II) are: 1) it is possible to use it as an open system that can operate under sink conditions which facilitates the dissolution of poorly water-soluble drugs as well as changing the dissolution medium within a range of physiological pH throughout the test [31]. 2) The flow-through cell method has a continuous extraction of the drug, simulating the absorption into the systemic circulation generating an intermittent flow of the dissolution medium into the cell where the dosage form is placed [32] and 3) USP apparatus IV better simulates the hydrodynamic environment that is found inside the gastrointestinal tract [33].
The flow-through cell generally gives better results than the membrane-type dissolution apparatus; therefore, the flow-through cell could be useful for predicting in vivo concentration curves from in vitro dissolution curves [21]. The mathematical treatment of dissolution data by numerical convolution is simple and offers an easy way to design bioequivalent drug products in the development stage [11], facilitating the development of multi-source formulations with better in vivo performance. The use of a USP apparatus IV is suggested to test the in vitro release behavior of indomethacin in rectal dosage forms.
The rectal route of administration is recommended for unconscious patients, those who have difficulty swallowing a solid dosage form, and for small children. Post-marketing in vitro dissolution studies of rectal formulations are essential to monitor the main quality attributes of the pharmaceutical drug product because different storage conditions can modify the in vitro release rate and, therefore, the absorption and the timely manifestation of the therapeutic effect. In addition, previous reports have shown a good prediction of the pharmacokinetic parameters of drugs with solubility problems (oral dosage forms) using dissolution data from the USP apparatus IV [34,35].
Dissolution studies are currently available in vitro laboratory resources that allow the determination of the release process of the drug from the formulation to be administered. These resources have some advantages, such as the ease of obtaining data under circumstances similar to those of the body, as well as avoiding expenses for in vivo absorption studies. Some disadvantages of in vivo performance predictions from dissolution data through mathematical calculations lie in inaccuracies due to variations in the pharmacokinetic data with which the calculations are generated or the scarcity of plasma data from a study in humans that allows validating the proposed predictions. In our study, no actual human data were freely available. In addition, the lack of training in data processing limits the widespread use of this type of prediction. One of the repercussions of this approach in humans is that these predictions do not consider the physiological conditions modified by diseases previously detected in patients; however, all changes in the formulations are made to improve absorption and promote the timely manifestation of the therapeutic effect.
The proposed simulations allow us to determine the appropriate in vitro conditions for evaluating the biopharmaceutical quality of indomethacin multisource rectal formulations through dissolution studies. Thus, these formulations can be safely interchangeable with reference drug products and can promote the development of therapeutic regimens at a low cost for patients.
CONCLUSION
Dissolution profiles of indomethacin rectal formulations were obtained in the hydrodynamic environment of USP apparatuses I and IV. After the experiments, similar in vitro release performance, regardless of the dissolution apparatus used and the addition of surfactant, was only obtained for the R drug product. For this formulation, values between 71% and 95% of the released drug were observed at 60 minutes. The best mathematical model to explain the drug-release mechanism is the Weibull function. On the other hand, the G formulation was more sensitive to the changes generated by the hydrodynamic environment and the addition of a surfactant. At 60 minutes, less than 29% of the dissolved drug (USP apparatus I without 1% SDS) and more than 81% (flow-through cell method with 1% SDS) were detected. The in vitro release of this formulation was better explained by the Makoid–Banakar model (USP apparatus I) and Weibull function (USP apparatus IV). Hypothetical plasma concentration-time profiles calculated with dissolution data of R drug product and a convolution approach produced Cmax and AUC0–inf values (2.39 and 8.46 µgh/ml, respectively), similar to those found in an in vivo study. Validation of this kind of prediction gave PE < 10% for both pharmacokinetic parameters. The flow-through cell method at 16 ml/min and phosphate buffer (0.1 M, pH 7.4) without 1% SDS were the best in vitro conditions to test multisource formulations given that dissolution data of R formulation and the convolution approach generated in vivo behavior as previously reported. Due to several characteristics of the flow-through cell method, especially hydrodynamic environment like that found in the gastrointestinal tract (generated by laminar flow and the design of the cell where the dosage form is placed), the production of in vitro data and their mathematical transformation in hypothetical in vivo behavior was easy to establish. Human studies with indomethacin suppositories are needed to confirm these findings.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to the concept and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the revision to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
FINANCIAL SUPPORT
There is no funding to report.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included in this research article.
USE OF ARTIFICIAL INTELLIGENCE (AI)-ASSISTED TECHNOLOGY
The authors declared that they have not used artificial intelligence (AI)-tools for writing and editing of the manuscript. No images were manipulated using AI.
PUBLISHER’S NOTE
All claims expressed in this article are solely those of the authors and do not necessarily represent those of the publisher, the editors, and the reviewers. This journal remains neutral regarding jurisdictional claims in published institutional affiliation.
REFERENCES
1. Thakur S, Riyaz B, Patil A, Kaur A, Kapoor B, Mishra V. Novel drug delivery systems for NSAIDs in management of rheumatoid arthritis: an overview. Biomed Pharmacother. 2018;106:1011–23. CrossRef
2. Patai A, Solymosi N, Mohácsi L, Patai AV. Indomethacin and diclofenac in the prevention of post-ERCP pancreatitis: a systematic review and meta-analysis of prospective controlled trials. Gastrointest Endosc. 2017;85(6):1144–56e1. CrossRef
3. Yang C, Zhao Y, Li W, Zhu S, Yang H, Zhang Y, et al. Rectal nonsteroidal anti-inflammatory drugs administration is effective for the prevention of post-ERCP pancreatitis: an update meta-analysis of randomized controlled trials. Pancreatology 2017;17(5):681–8. CrossRef
4. Nascimento ALCS, Fernandes RP, Charpentier M, Ter Horst JH, Caires FJ, Chorillo M. Co-crystals of non-steroidal anti-inflammatory drugs (NSAIDs): insight toward formation, methods, and drug enhancement. Particuology 2021;58(3):227–41. CrossRef
5. Nascimento ALCS, Martins ICB, Spósito L, Morais-Silva G, Duarte JL, Rades T, et al. Indomethacin-omeprazole as therapeutic hybrids? Salt and co-amorphous systems enhancing physicochemical and pharmacological properties. Int J Pharm. 2024;653:123857. CrossRef
6. Mohammed A, Elshaer A, Sareh P, Elsayed M, Hassanin H. Additive manufacturing technologies for drug delivery applications. Int J Pharm. 2020;580:119245. CrossRef
7. Aiache JM, Islasse M, Beyssac E, Aiache S, Renoux R, Kantelip JP. Kinetics of indomethacin release from suppositories. In vitro-in vivo correlation. Int J Pharm. 1987;39(3):235–42. CrossRef
8. De Muynck C, Remon JP. Influence of fat composition on the melting behavior and on the in vitro release of indomethacin suppositories. Int J Pharm. 1992;85(1–3):103–12. CrossRef
9. Uzunkaya G, Bergi?adi N. In vitro drug liberation and kinetics of sustained release indomethacin suppository. Il Farmaco. 2003;58(7):509–12. CrossRef
10. United States Pharmacopeia 47/National Formulary 42. United States Pharmacopoeial Convention, Inc; 2024.
11. Hassan HA, Charoo NA, Ali AA, Alkhatem SS. Establishment of a bioequivalence-indicating dissolution specification for candesartan cilexetil tablets using a convolution model. Dissol Technol. 2015;22(2):36–43. CrossRef
12. Emara LH, El-Menshawi BS, Estefan MY. In vitro-in vivo correlation and comparative bioavailability of vincamine in prolonged-release preparations. Drug Dev Ind Pharm. 2000;26(3):243–51. CrossRef
13. Jinno J, Kamada N, Miyake M, Yamada K, Mukai T, Odomi M, et al. In vitro-in vivo correlation for wet-milled tablet of poorly water-soluble cilostazol. J Control Rel. 2008;130(1):29–37. CrossRef
14. Xie F, Ji S, Cheng Z. In vitro dissolution similarity factor (f2) and in vivo bioequivalence criteria, how and when do they match? Using a BCS class II drug as a simulation example. Eur J Pharm Sci. 2015;66:163–72. CrossRef
15. Zhang Y, Huo M, Zhou J, Zou A, Li W, Yao C, et al. DDSolver: an add-in program for modeling and comparison of drug dissolution profiles. AAPS J. 2010;12(3):263–71. CrossRef
16. Yuksel N, Kanik AE, Baykara T. Comparison of in vitro dissolution profiles by ANOVA-based, model-dependent and -independent methods. Int J Pharm. 2000;209(1–2):57–67. CrossRef
17. Zhang Y, Huo M, Zhou J, Xie S. PKSolver: an add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput Methods Programs Biomed. 2010;99(3):306–14. CrossRef
18. Qureshi SA. In vitro-in vivo correlation (IVIVC) and determining drug concentrations in blood from dissolution testing – a simple and practical approach. Open Drug Deliv J. 2010;4:38–47. CrossRef
19. Rastogi V, Yadav P, Lal N, Rastogi P, Singh BK, Verma N, et al. Mathematical prediction of pharmacokinetic parameters-an in-vitro approach for investigating pharmaceutical products for IVIVC. Future J Pharm Sci. 2018;4(2):175–84. CrossRef
20. Efentakis M, Al-Hmoud H, Buckton G, Rajan Z. The influence of surfactants on drug release from a hydrophobic matrix. Int J Pharm. 1991;70(1–2):153–8. CrossRef
21. Lootvoet G, Beyssac E, Shiu GK, Aiache JM, Ritschel WA. Study on the release of indomethacin from suppositories: in vitro-in vivo correlation. Int J Pharm. 1992;85(1–3):113–20. CrossRef
22. Azarmi S, Roa W, Löbenberg R. Current perspectives in dissolution testing of conventional and novel dosage forms. Int J Pharm. 2007;328(1):12–21. CrossRef
23. Podczeck F. Comparison of in vitro dissolution profiles by calculating mean dissolution time (MDT) or mean residence time (MRT). Int J Pharm. 1993;97(1–3):93–100. CrossRef
24. Archondikis A, Papaioannou G. Comparative study of two dissolution methods for indomethacin suppositories from fatty and water-soluble bases. Int J Pharm. 1989;55:217–20. CrossRef
25. Ilango KB, Kavimani S. A systematic review of the mathematical models of pharmaceutical dosage forms. Int J Curr Pharm Rev Res. 2015;6(1):59–70.
26. Medina JR, Salazar K, Hurtado M, Cortés AR, Domínguez-Ramírez AM. Comparative in vitro dissolution study of carbamazepine immediate-release products using the US paddles method and the flow-through cell system. Saudi Pharm J. 2014;22:141–7. CrossRef
27. Fang JB, Robertson VK, Rawat A, Flick T, Tang ZJ, Cauchon NS, et al. Development and application of a biorelevant dissolution method using USP apparatus 4 in early phase formulation development. Mol Pharmaceut. 2010;7(5):1466–77. CrossRef
28. De Muynck C, Lefebvre RA, Remon JP. Study of the bioavailability of four indomethacin suppository formulations in healthy volunteers. Int J Pharm. 1994;104(1):87–91. CrossRef
29. Xu J, Zhang L, Shao X. Application of bio-predictive dissolution tools for the development of solid oral dosage forms: current industrial experience. Drug Dev Ind Pharm. 2022;48(3):79–97. CrossRef
30. Paprská?ová A, Možná P, Oga EF, Elhissi A, Alhnan MA. Instrumentation of flow-through USP IV dissolution apparatus to assess poorly soluble basic drug products: a technical note. AAPS PharmSciTech. 2016;17(5):1261–5. CrossRef
31. Fotaki N. Flow-through cell apparatus (USP apparatus 4): operation and features. Dissolut Technol. 2011;18(4):46–9. CrossRef
32. Todaro V, Persoons T, Grove G, Healy MA, D’Arcy DM. Characterization and simulation of hydrodynamics on the paddle, basket and flow-through dissolution testing apparatuses – a review. Dissolut Technol. 2017;24(3):24–36. CrossRef
33. Abdelfattah F, Taha N, Abdou A, Mursi N, Emara L. Prediction of in vivo performance of ibuprofen immediate-release products using different dissolution models. J Appl Pharm Sci. 2022;12(8):193–201. CrossRef
34. Medina-López JR, Lugo-Ortíz R, Contreras-Jiménez JM, Hurtado M, Helmy SA. Dissolution performance of verapamil-HCl tablets using USP Apparatus 2 and 4: prediction of in vivo plasma profiles. Dissol Technol. 2023;30(4):230–7. CrossRef
35. Medina-López R, Vera-Ángeles YA, Reyes-Ramírez FD. Simulation of indomethacin plasma levels: influence of the hydrodynamics of the USP basket apparatus and flow-through cell system to evaluate capsules. Lat Am J Pharm. 2025;44(2):182–8.