INTRODUCTION
The global healthcare landscape grapples with chronic bacterial infections, resulting in prolonged patient suffering and inflated healthcare expenses. These infections profoundly impact patients’ quality of life and necessitate effective treatment strategies [1,2]. In 2017 and 2019, the Indian Council of Medical Research reported several pathogens that were found to be responsible for such maladies. These findings have been corroborated by the World Health Organization and the Center for Disease Control and Prevention, signifying the escalating necessity to address this global health threat [2–5].
Among the arsenal of antibiotics utilized to combat chronic bacterial infections, meropenem trihydrate (MPN) is a broad-spectrum and last-resort antibiotic option [6]. Its effectiveness against a vast range of bacteria designates it as an invaluable therapeutic agent for managing severe infections. Currently, MPN is marketed as an intravenous formulation with various doses in the treatment regimen. However, the quest for optimization has driven research toward nanoformulations such as meropenem-loaded solid lipid nanoparticles [7], liposomes [8], and mesoporous silica nanoparticles [6]. These innovations promise enhanced drug delivery and heightened treatment efficacy [9,10].
The accurate and precise quantification of MPN in dosage forms is paramount for elucidating its dose-dependent therapeutic action. Various analytical methods have been reported for MPN quantification, including ultraviolet (UV) spectrometry [11], high-performance liquid chromatography-ultraviolet spectrometry (HPLC-UV) [11–15], and liquid chromatography-mass spectrometry (LC-MS) [15,16]. Despite the availability of these methodologies, high-performance liquid chromatography (HPLC) with UV detection is often the preferred choice for quantifying drugs in pharmaceutical formulations, credited for its rapidness, robustness, reliability, selectivity, and sensitivity, which surpass traditional UV techniques [17–19]. Additionally, the lower cost and simpler instrumentation of HPLC-UV compared to LC-MS make it more accessible for a wide range of analytical laboratories. Therefore, we used the HPLC-UV method in the current investigation to quantify MPN [11,12]. However, a review of the literature reveals significant limitations within reported methods for estimating MPN. These shortcomings include poor sensitivity [14,20,21], excessive use of organic solvents that are not environmentally friendly and lead to high analysis costs [14,20–22], long run times that render the process time-intensive [11,23–25], complex method of elution [26–28], complicated mobile phase preparations [29], the complex and costly use of detectors such as mass spectrometry [26–28], incomplete method validation [14,20–28,30–33], and a notable absence of a reliable analytical HPLC method for the estimation of MPN. The reported analytical methods are lacking in providing comprehensive details of the impact of various factors as well as the interaction effect of factors on responses [11,14,20–30,31–33]. Additionally, there is a dearth of methods for quantifying the MPN in the marketed formulation and novel nanoformulation (nanosponges). Hence, the pressing need remains to develop a robust and validated HPLC-UV analytical method to quantify MPN in pharmaceutical formulations. The detailed drawbacks of existing methods are summarized in Table 1 below, highlighting the novelty and necessity of this study to address the gaps in current research and methodology.
 | Table 1. Comparison of the proposed analytical method with reported methods. [Click here to view] |
Systematic evaluation of factors affecting analytical method performance is essential but challenging. This study employs a quality by design (QbD) approach to method development, focusing on identifying and controlling critical factors to ensure robustness, reliability, accuracy, and precision. The QbD approach also minimizes variability and enhancing method performance [34–37]. The objective is to develop an HPLC-UV method for quantifying MPN in pharmaceutical formulations using this systematic framework.
This study also examines the stability and degradation behavior of MPN under stress conditions to identify degradation pathways and stability concerns, ensuring the safety and efficacy of pharmaceutical formulations. Additionally, it addresses gaps in the literature by analyzing MPN quantification in marketed formulations and innovative nanosponges.
Growing awareness of environmental impacts from chemicals underscores the need for sustainable practices aligned with the United Nations’ Sustainable Development Goals (SDGs), particularly SDG 12: “Responsible Consumption and Production”. These efforts also support SDG 3: “Good Health and Well-being” and SDG 14: “Life Below Water” by minimizing harmful chemical effects, and driving eco-friendly analytical methodologies [38,39].
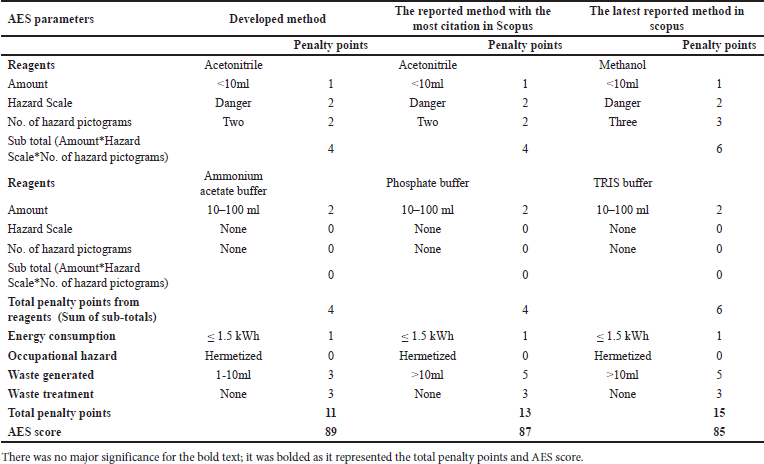
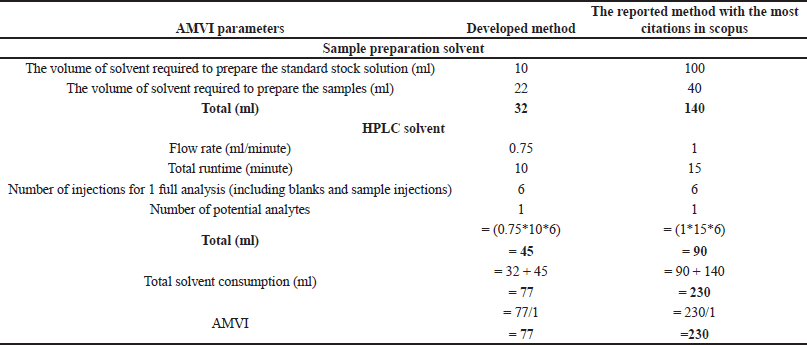
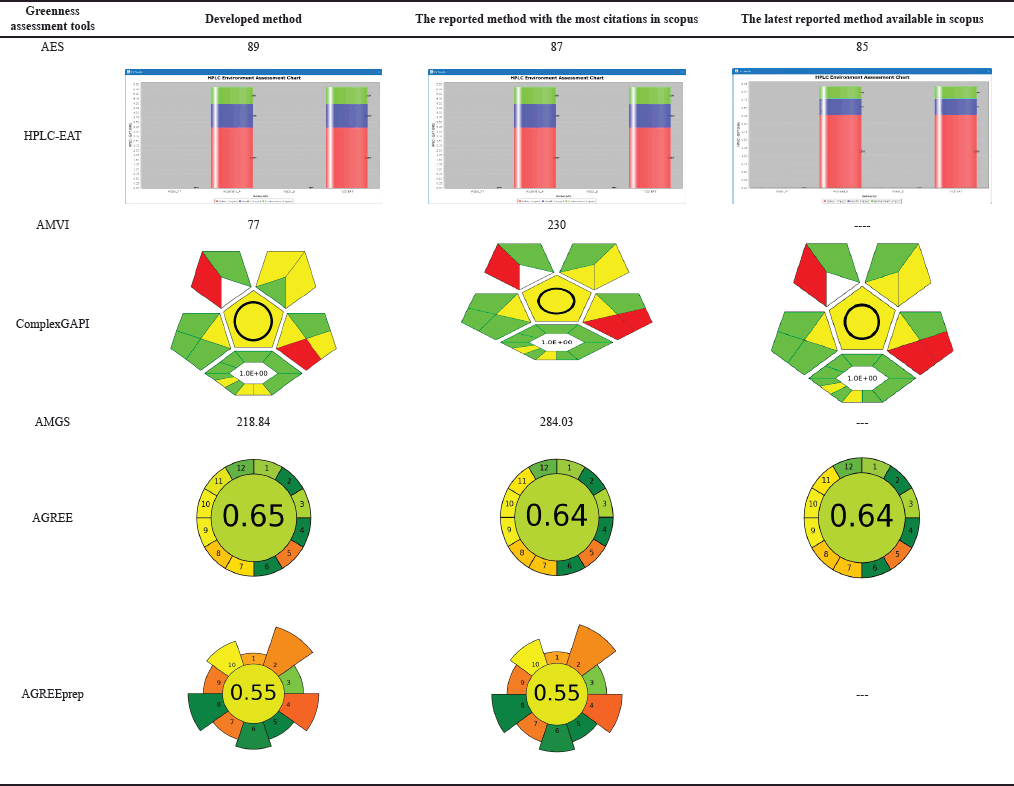
Green analytical chemistry (GAC) promotes sustainable analytical practices that balance accuracy, robustness, and environmental stewardship [40,41]. This paradigm shift prioritizes energy efficiency, waste reduction, and minimal environmental impact in liquid chromatography, aligning with SDG 12 and related goals. Advanced greenness evaluative tools, from Analytical Eco-Scale (AES) to the analytical greenness (AGREE) calculator, provide comprehensive assessments of analytical methods, supporting a synergistic evaluation approach [42,43]. Our developed method was compared against the existing analytical methods (“reported method with most citations in Scopus” [11] and the “latest reported method available in Scopus” [14]). These methods were chosen due to their widespread acclaim and the most up-to-date information available in the scientific community. By juxtaposing our developed method with these highly regarded counterparts, we aimed to demonstrate its sustainable essence and competitive prowess within existing analytical approaches.
This research advances analytical methodologies for MPN quantification, providing insights into its stability and degradation behavior. Our greenness assessment highlights the method’s minimal environmental impact, achieved through advanced tools, and sustainable practices. These efforts contribute to developing MPN-based formulations for chronic bacterial infections while promoting eco-friendly analytical research.
MATERIALS
MPN (purity ≥98%) was procured from Sigma Aldrich. The buffer salts utilized in this research, including ammonium acetate (purity ≥97%), ammonium formate (purity 97%), and sodium acetate (purity ≥99%), were procured from Merck Life Science Pvt. Ltd. (Mumbai, India). Potassium dihydrogen phosphate (purity, min. 99%) was from Himedia Labs Pvt. Ltd. (Mumbai, India), and sodium hydroxide pellets (purity, min. 97%) were from Finar Ltd. (Gujarat, India). The HPLC-grade solvents of acetonitrile (purity, min 99.9%), hydrochloric acid (37%), hydrogen peroxide (30%), glacial acetic acid (100%), and orthophosphoric acid (purity, min 85%) were procured from Merck Life Science Pvt. Ltd. (Mumbai, India). Methanol (purity, min. 99.80%), triethylamine LR (purity, min. 99.00%), and formic acid (purity, 85%) were purchased from Finar Ltd. (Gujarat, India). Beta-cyclodextrin (β-CD) was procured as ex-gratis from Ashland, India, and diphenyl carbonate (DPC) was purchased from Spectrochem, Pvt. Ltd. (Mumbai, India) for nanosponges preparation. Ferric chloride (purity ≥99%) and acetone (purity, 99% min) were procured from Merck Life Science Pvt. Ltd. (Mumbai, India).
INSTRUMENTATION
The present research study employed the LC-2010C HT, a HPLC instrument manufactured by Shimadzu, Japan. This advanced device is equipped with a dual-wavelength UV detector, a column oven, and an autosampler, all provided by Shimadzu. To record and analyze the chromatographic data, LC Solution software version 5.57 was utilized. The various analytical columns used are Kinetex C18 (250 mm*4.6 mm*5 μ) column from Phenomenex, USA; Hyperclone C18 (250 mm*4.6 mm*5 μ) column from Phenomenex; Chromasol Jade C18 (250 mm*4.6 mm*5 μ) column from Intek; and Chromasol Onyx C18 (250 mm*4.6 mm*5 μ) column from Intek.
The mobile phase was filtered through a 0.22-micron cellulose nitrate membrane using a Millipore glass vacuum filtration unit and sonicated for 10 minutes in a GT Sonic Ultrasonic Cleaner (Servewell Instruments, India). pH measurements were performed with a Systronics Micro-Controller-based pH system 361, ensuring precise control.
METHOD DEVELOPMENT
QbD approach
The study adopted the Analytical QbD framework to develop a robust and reproducible HPLC-UV method for estimating MPN in marketed and nanosponges formulations, aligning with International Conference on Harmonisation (ICH) Q2(R1), Q8(R2), and Q14 guidelines [44–46].
Defining the analytical target profile (ATP) and determining critical performance attributes (CPAs)
The ATP was defined to ensure method performance. CPAs were identified based on prior knowledge, literature review, and preliminary trials [35].
Risk assessment
A systematic risk assessment was performed to evaluate the impact of Critical Method Attributes (CMAs) and Critical Procedure Parameters (CPPs) on CPA. This risk assessment was conducted using an Ishikawa Fishbone Diagram (Fig. 1) to identify potential risks associated with method and procedure attributes. Preliminary trials conducted using a one-factor-at-a-time (OFAT) design, allowed for the identification of critical factors affecting method performance [47].
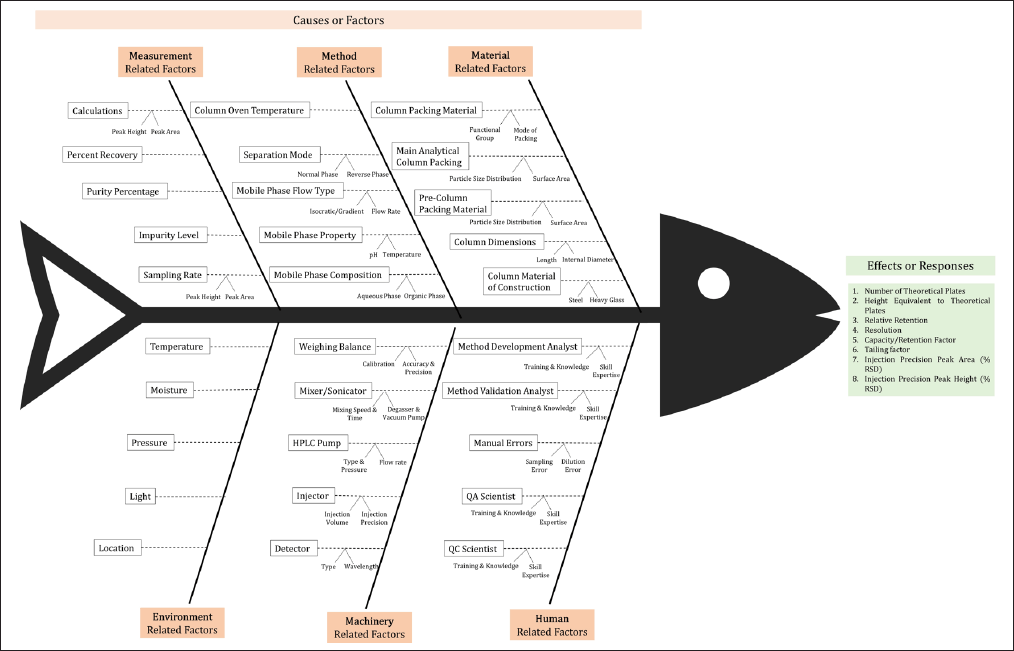 | Figure 1. Identification of risk factors by the Ishikawa Fishbone diagram. [Click here to view] |
Preparation of the mobile phase and standard solutions
The mobile phase was prepared by dissolving HPLC-grade ammonium acetate in 1,000 ml of MilliQ water and adjusting to the desired pH of 5.1. A 1,000 μg/ml standard stock solution of MPN was prepared by dissolving 10 mg of MPN in the mobile phase, with subsequent dilutions to achieve the required concentrations.
Design of experiments (DoEs)
DoE principles were applied to optimize CMAs and CPPs. Screening and optimization experiments were conducted to evaluate the effect of independent factors on dependent variables using Design-Expert software (v9.0.5.1, Stat-Ease Inc., USA).
Screening of independent factors using fractional factorial design
A two-level fractional factorial design (2V5-1) with three center points and triplicates was employed to screen five independent factors (Table 2) based on their main and interaction effects. The design suggested 51 experiments, enabling efficient factor prioritization. Factor levels were chosen based on literature and preliminary studies [48].
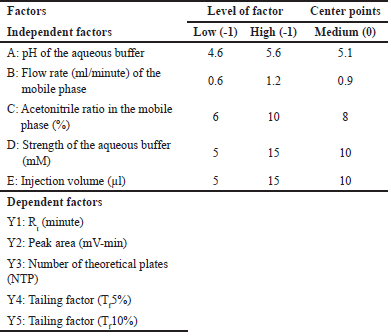 | Table 2. Independent factors with its level, center points, and their dependent factors in 2V (5-1) fractional factorial design. [Click here to view] |
Optimization of factors using Box-Behnken design
Following the screening phase, the three most significant factors were optimized using the Box-Behnken design, an economical response surface methodology suitable for optimizing three independent factors. This design estimates the main and second-order interaction effects of input factors on responses with minimal experimentation. The software suggested 12 experiments, and the optimum levels of the factors were identified through these trials.
Development and verification of the method operable design region (MODR)
The MODR defines a flexible multidimensional space where the interaction of independent factors ensures minimal variability [35]. MODR was established through numerical optimization, with specified limits for all individual responses.
Numerical optimization identified the optimal solution with the highest desirability score. The MODR was further validated through three confirmatory experimental runs, and the results were consistent with model-predicted values.
VALIDATION OF THE OPTIMIZED HPLC METHOD
Validation was conducted according to ICH Q2(R1) guidelines to assess the method’s performance for its intended use [49].
Specificity
Specificity was evaluated by injecting replicates of blank (diluent), MPN standard (5 μg/ml), marketed formulation (equivalent to 10 μg/ml of MPN), placebo, and novel nanosponges formulation (equivalent to 10 μg/ml of MPN). Chromatograms were analyzed for potential interference from excipients or diluents at MPN’s retention time (Rt).
Linearity
The method’s linearity was tested using MPN concentrations of 0.1, 0.25, 0.5, 1, 2.5, 5, 7.5, 10, and 12.5 μg/ml. Each concentration was injected in quintuplicate, and a linear regression graph was plotted between the concentration (μg/ml) and peak area (mV-min). The regression equation’s slope and intercept were calculated [50].
Accuracy and precision
Accuracy was assessed by injecting three concentrations (4, 5, and 6 μg/ml) in sextuplicate. It was evaluated by analyzing these concentrations, and the mean percentage recovery was calculated for each concentration [50].
Precision was assessed using four quality control (QC) concentrations: quantitation limit (QL), lower QC (3× QL), middle QC (average of lower and higher QC), and higher QC (70% of the highest linearity concentration). Intra-day precision was evaluated by analyzing sextuplicates of the QC concentrations at two different time points within the same day (09:00 and 21:00). Inter-day precision was assessed by performing duplicate injections of the QC concentrations over three consecutive days. For both intra-day and inter-day analyses, the peak area and percentage relative standard deviation (%RSD) were calculated to determine precision.
Statistical comparison with a reported method to validate our method’s performance, we compared it with the previously reported method by Mendez et al. [11] using two statistical tests [11].
Comparison of method precision (F-test)
The F-test was used to compare how precise both methods are by comparing their variations in measurements. This tcresults than the other.
Comparison of method accuracy (t-test)
Student’s t-test was used to compare the accuracy between both methods by comparing their mean recovery values. This helps determine if both methods give similar results when measuring the same thing. Both statistical tests were performed at a 95% confidence level. If the calculated values were less than the critical values from standard statistical tables, it would indicate no significant difference between the methods.
Sensitivity
The detection limit (DL) and QL were calculated using the following formulas:
where σ = the residual standard deviation of the regression line
S = slope of the regression line.
Robustness
Robustness was evaluated by analyzing the impact of minor variations in experimental conditions (Table 3). An MPN concentration of 5 μg/ml was injected thrice under altered conditions, and parameters such as peak area, Rt, Tf5%, Tf10%, and NTP were monitored for %RSD.
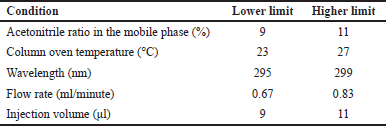 | Table 3. Different conditions for robustness studies. [Click here to view] |
System suitability
System suitability was confirmed by injecting 5 μg/ml of MPN in sextuplicate. Parameters such as peak area, Rt, Tf5%, Tf10%, and NTP were calculated along with %RSD to ensure the system met the required criteria.
BENCHTOP STABILITY STUDIES OF THE MPN SOLUTION
The stability and degradation studies for MPN were conducted to evaluate its behavior under various conditions. Benchtop stability of the MPN solution was assessed using two concentrations, 4.59 μg/ml (lower QC) and 8.75 μg/ml (higher QC). Fresh solutions were prepared and stored at ambient temperature over three days, labeled D1, D2, and D3. On Day 3, all solutions were analyzed in triplicate. The mean peak areas of D1 (48 hours old) and D2 (24 hours old) were compared to D3 (freshly prepared) to calculate the percentage stability and similarity indices.
AUTOSAMPLER POST-OPERATIVE STABILITY STUDIES OF MPN SOLUTION
Autosampler post-operative stability studies were conducted to determine the integrity of MPN stored at 4°C in autosampler vials for 24 hours. The lower and higher QC concentrations were analyzed in triplicate and compared to freshly prepared samples. Stability percentages and similarity indices were calculated based on peak area comparisons [51].
Similarity index for autosampler postoperative stability
BENCHTOP STABILITY STUDIES OF THE MOBILE PHASE
The benchtop stability of the mobile phase was assessed by preparing and storing it at ambient temperature for three consecutive days (labeled M1, M2, and M3). On Day 3, a 5 μg/ml MPN solution was analyzed using all three mobile phases in triplicate, with M1 (48 hours old) and M2 (24 hours old) compared to M3 (freshly prepared).
Similarity index for 48 hours stability
Similarity index for 24 hours stability
DEGRADATION STUDIES
Degradation studies examined MPN under stress conditions, including acid, alkali, oxidative, and photolytic exposures. Acid-induced degradation was conducted under real-time and accelerated conditions by treating MPN (5 μg/ml) with 0.1 N and 1 N HCl. Samples were stored at ambient temperature for 24 hours or at 60°C for 6 hours, then neutralized with NaOH and analyzed. Alkali-induced degradation was performed similarly, using 0.1 N and 1 N NaOH for treatment and subsequent neutralization with HCl. Oxidative degradation involved treating MPN with 3% H2O2, either stored at ambient temperature for 24 hours or at 60°C for 6 hours, followed by analysis. Photolytic degradation was evaluated by exposing MPN to UV light (direct sunlight) for 24 hours, and the resulting degradation products were analyzed. This comprehensive assessment of MPN’s stability and degradation under varying conditions provides insights into its robustness and suitability for analytical applications [51].
APPLICATION OF THE DEVELOPED ANALYTICAL METHOD FOR QUANTIFICATION OF MPN IN MARKETED PRODUCT AND NOVEL NANOFORMULATIONS
The developed HPLC method is considered a valuable tool only after its practical application to quantify MPN in dosage forms. MPN was analyzed in the marketed powder for injection formulation (Merosure 125 mg, Alkem Laboratories, India) and the novel β-CD nanosponges formulation to test the practical feasibility of the method.
Quantification of MPN in the marketed formulation
The validated method was used to analyze and quantify the MPN in the marketed powder for injection formulation. The marketed formulation’s standard stock solution was prepared by accurately weighing 5.45 mg (equivalent to 5 mg of MPN), transferring it to a 10 ml volumetric flask, and filling the volume with the mobile phase at a final concentration of 500 μg/ml. Further dilutions were done with the mobile phase to obtain the desired concentration (10 μg/ml). The validated method was used to analyze the desired concentration, and the Rt and peak area were measured. The amount of MPN in the desired concentration was calculated by comparing the peak area [35].
Quantification of MPN in β-CD-nanosponges
Preparation of β-CD cross-linked nanosponges
β-CD nanosponges were prepared using the melt method, involving a cross-linking reaction between β-CD and DPC in a 1:4 weight ratio. The homogenously triturated mixture was reacted for 5 hours at 100°C and left overnight at ambient temperature. The resulting cross-linked nanosponges were purified via Soxhlet extraction using acetone, and the phenol by-product was removed. The purified nanosponges were dried at 50°C and tested with 1% ferric chloride solution to ensure phenol-free purity, resulting in a pure white powder stored at 25°C for future use [52].
Preparation of MPN encapsulated β-CD-nanosponges
MPN encapsulation into nanosponges was achieved using a solvent immersion technique. A 1:1 ratio of MPN and nanosponges was used, where the nanosponges were dispersed in water and sonicated with a probe sonicator (20% amplitude for 10 minutes with 2-second breaks every 10 seconds). MPN was then added to the dispersion, stirred for 6 hours, and centrifuged at 2,500 rpm for 10 minutes. The colloidal supernatant containing drug-encapsulated nanosponges was collected, freeze-dried, and stored for further analysis [52].
Encapsulation efficiency
To determine encapsulation efficiency, MPN-encapsulated nanosponges were weighed and dispersed in methanol, followed by sonication to break the nanosponges. The sonicated mixture was centrifuged, and the supernatant was diluted with the mobile phase for HPLC analysis. The Rt and peak areas were recorded, and the amount of MPN was calculated by comparing the peak areas with the standard. Encapsulation efficiency was calculated using the below-mentioned formulas [52].
STATISTICAL ANALYSIS
All the data obtained from the experiments were subjected to statistical analysis. Results are presented as mean values and their respective % RSD. Statistical significance was determined at a 95% confidence level.
RESULT AND DISCUSSION
Defining an ATP and determining CPAs
The ATP was established to ensure a robust and reliable HPLC-UV method for quantifying MPN, addressing systematic and inherent variability while ensuring system suitability. As part of the QbD approach, CPAs were identified to significantly impact the method’s performance. They are (a) Rt (min), (b) peak area (mV-min), (c) tailing factor, and (d) number of theoretical plates.
Risk assessment
A risk assessment was conducted using an Ishikawa fishbone diagram to identify the CMAs and CPPs that could influence the analytical method. Preliminary trials (Table 4), based on an OFAT design, were conducted, and the variables such as buffer pH, flow rate, acetonitrile ratio in the mobile phase, buffer strength, and injection volume were identified as critical based on their significant impact on method performance.
 | Table 4. Preliminary trial for the identification of risk factors. [Click here to view] |
Selection of the mobile phase
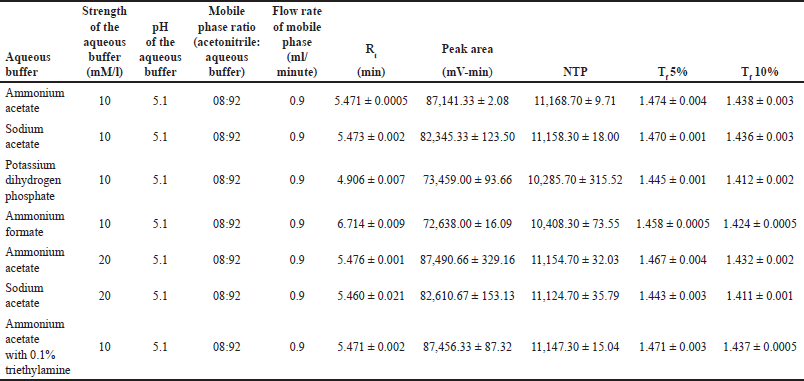
Acetonitrile was chosen as the organic mobile phase because it has a higher vapor pressure than methanol and a lower internal pressure in the column, leading to better efficiency and column lifetime. The aqueous mobile phase composition was optimized by evaluating the buffer type, pH, and concentration. Considering MPN’s pKa values (2.9 and 7.4), a buffer pH of 5.1 was selected to maintain the drug in a consistent ionized or unionized state without compromising the silica column integrity.
Four buffer systems (ammonium acetate, ammonium formate, sodium acetate, and potassium dihydrogen phosphate) were screened at a concentration of 10 mM with a mobile phase ratio of 92:08 (aqueous:organic). Ammonium acetate demonstrated superior performance, yielding a peak area of 87,141.33 ± 2.08 mV-min and 11,168.70 ± 9.71 theoretical plates, compared to sodium acetate with 82,345.33 ± 123.50 mV-min and 11,158.30 ± 18.00 theoretical plates.
Selection of the stationary phase
In continuation of the OFAT design, various stationary phases, including the Phenomenex Kinetex C18 (250 mm*4.6 mm*5 μ) column, Phenomenex Hyperclone C18 (250 mm*4.6 mm*5 μ) column, Intek Chromasol Jade C18 (250 mm*4.6 mm*5 μ) column, and Intek Chromasol Onyx C18 (250 mm*4.6 mm*5 μ) column, were screened.
Acetonitrile and 10 mM ammonium acetate in a ratio of 8:92 were used in the preliminary trials for the stationary phase selection. An MPN concentration of 5 μg/ml was prepared and analyzed in the four stationary phases.
Among the four stationary phases, the Phenomenex Kinetex C18 column significantly showed consistent results with a better peak area, and therefore, it was selected as the ideal stationary phase for the method development.
Design of experiments
Screening of independent factors using fractional factorial design
Screening of 5 independent factors [Factor A: pH of the buffer; Factor B: flow rate; Factor C: acetonitrile ratio in the mobile phase; Factor D: strength of the buffer; and Factor E: injection volume] was carried out using a fractional factorial design. Dependent responses were measured for all experimental runs, as tabulated in Table 5, and the relationship between factors and their respective responses was analyzed using graphical effects analysis, specifically Pareto charts (Fig. 2).
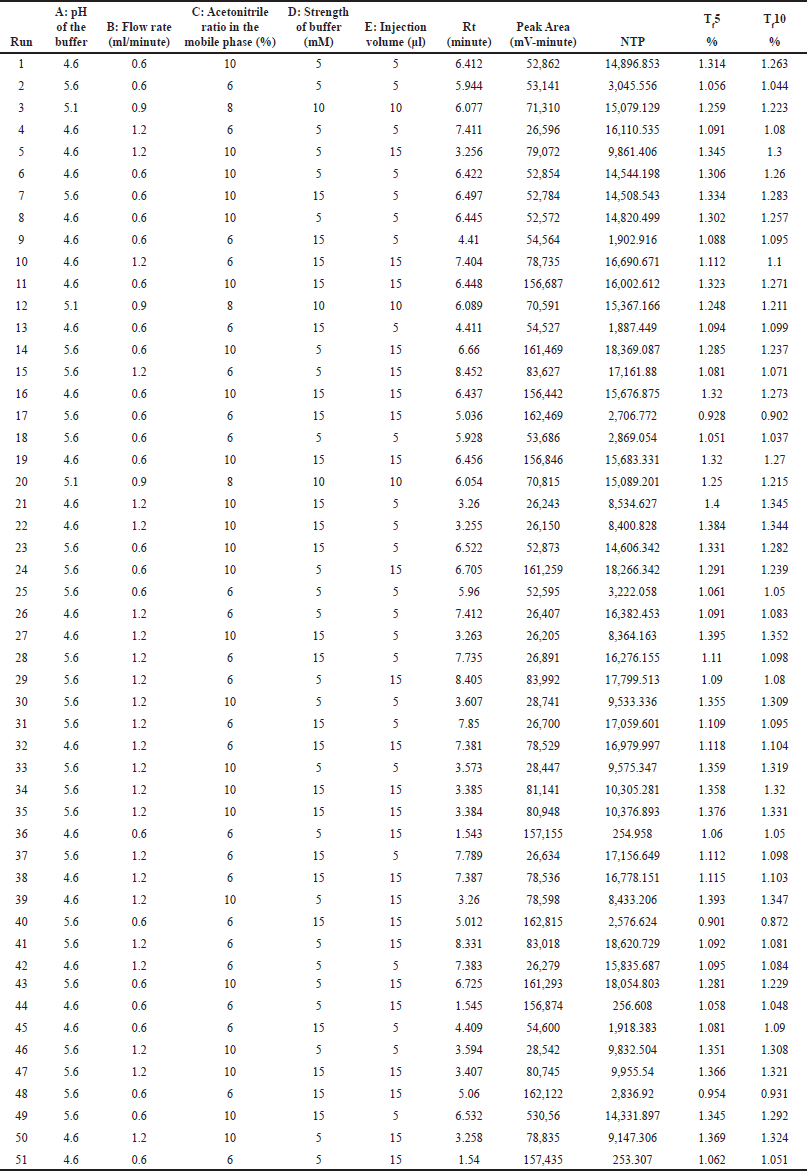 | Table 5. Experimental runs and their results based on fractional factorial design. [Click here to view] |
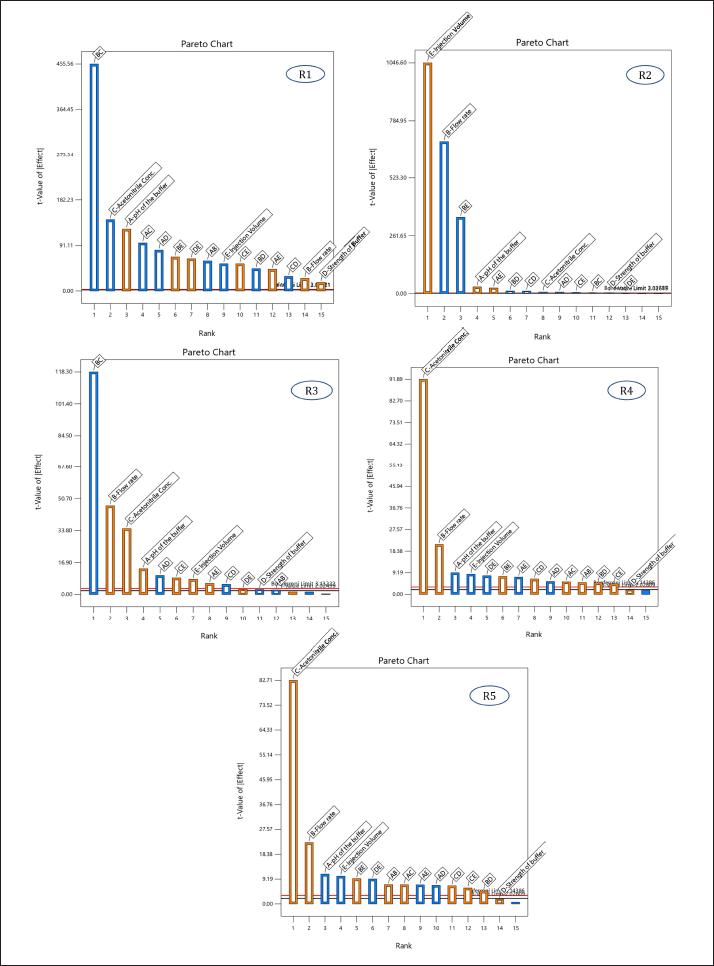 | Figure 2. Pareto chart illustrating the effect of independent variables [(A) pH of the buffer, (B) flow rate, (C) acetonitrile ratio in the mobile phase, (D) strength of the buffer, and (E) injection volume] on respective responses [(R1) Rt of MPN, (R2) peak area of MPN, (R3) number of theoretical plates, (R4) tailing factor 5%, and (R5) tailing factor 10%]. [Click here to view] |
The Pareto charts revealed that factors A, B, and C had t-values exceeding their Bonferroni limits, confirming their significance. Conversely, factors D and E were deemed non-significant due to their t-values falling below the Bonferroni limits.
Subsequently, numerical optimization was performed to determine the ideal values for the non-significant factors (buffer strength and injection volume) to achieve the desired responses. Among the potential solutions offered by the software, the solution with the highest desirability (desirability = 1) was chosen. As per the numerical optimization, the ideal injection volume was 10 μl, and the ideal buffer strength was 10 mM.
The factors A, B, and C were found significant and subjected to optimization using the Box-Behnken design.
Optimization of factors using Box-Behnken design
A 2-level Box-Behnken design with their center points was performed for three independent factors [A: pH of the buffer, B: flow rate, and C: acetonitrile ratio in the mobile phase]. The software recommended 12 experimental runs were performed, and the results are tabulated in Table 6. The effect of individual independent factors on responses was analyzed using a quadratic regression model through analysis of variance (ANOVA). The results of the ANOVA test for all the independent factors from the quadratic model represent the main effects and interaction effects of factors on responses, and the results are tabulated in Table 7. The impact of individual factors on responses was briefly discussed in a further section.
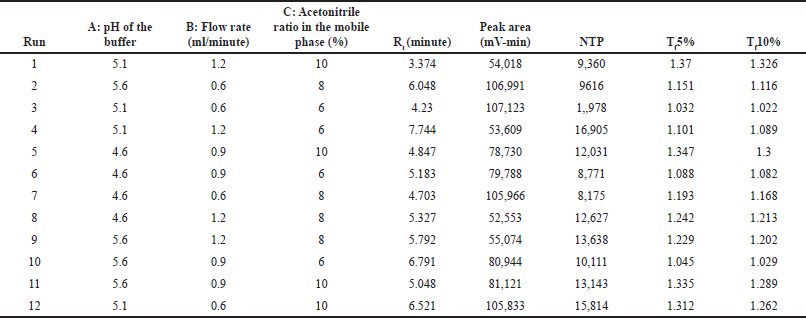 | Table 6. Experimental runs and their results based on Box-Behnken design. [Click here to view] |
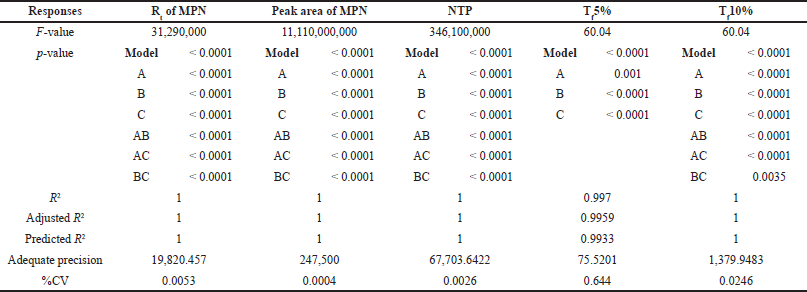 | Table 7. Results of the ANOVA test for all the independent factors from the quadratic model. [Click here to view] |
Analysis of the effect of individual independent factors on the Rt
The quadratic model and ANOVA results led to the following coded equation for Rt (Y1):
This equation, alongside Figure 3(R1), suggests that the buffer pH and flow rate positively affect Rt, while the acetonitrile ratio in the mobile phase negatively impacts it. At pH 4.6, MPN becomes more polar, and as the pH increases, its lipophilicity rises, enhancing retention. Increasing the acetonitrile ratio in the mobile phase reduces Rt due to decreased polarity. The flow rate also influences Rt, with a higher flow rate resulting in a slight increase in retention due to the interaction with acetonitrile concentration.
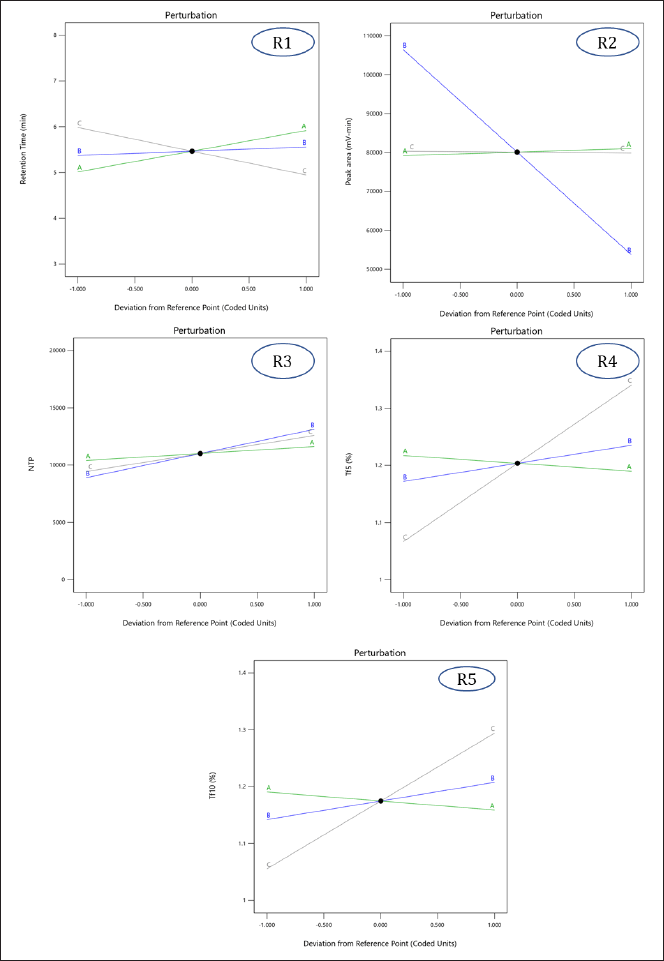 | Figure 3. Perturbation plots demonstrating the effect of independent variables [(A) pH of the buffer, (B) flow rate, and (C) acetonitrile ratio in the mobile phase] on individual responses [(R1) Rt of MPN, (R2) peak area for MPN, (R3) number of theoretical plates, (R4) tailing factor 5%, and (R5) tailing factor 10%]. [Click here to view] |
Analysis of the effect of individual independent factors on the peak area
The coded equation for peak area (Y2) is:
Here, buffer pH positively impacts peak area, while flow rate and acetonitrile ratio negatively affect it. Increasing the buffer pH enhances the degree of ionization, slightly increasing the peak area. Higher flow rates reduce peak area due to decreased analyte Rt, while an increased acetonitrile ratio causes a negligible decrease in peak area due to changes in ionization (Fig. 3(R2)).
Analysis of the effect of individual independent factors on the number of theoretical plates
The coded equation for the number of theoretical plates (Y3) is:
Flow rate, buffer pH, and acetonitrile ratio all positively affect theoretical plates. Contrary to theoretical understanding, increasing the flow rate resulted in a higher number of theoretical plates due to interactions with other factors. Increased pH enhances ionization, improving elution efficiency, and an increased acetonitrile ratio improves elution efficiency, increasing the number of theoretical plates (Fig. 3(R3)).
Analysis of the effect of individual independent factors on the tailing factor 5% and the tailing factor 10%
Equations 4 and 5 are coded equations for the tailing factor 5% (Y4) and the tailing factor 10% (Y5), respectively.
Flow rate and acetonitrile ratio increase the tailing factor, as higher flow rates reduce analyte-column interaction time, causing band broadening and peak tailing. Increased acetonitrile reduces polarity, resulting in broader peaks and higher tailing factors. Buffer pH, conversely, improves peak symmetry by altering the ionization state, and reducing tailing (Fig. 3 (R4) and (R5)).
Development and verification of method operable design space
After using a regression model to analyze the factors, numerical optimization was used to develop the design space. The software generates 47 prospective solutions, of which the optimized solution was chosen (Fig. 4.) based on the highest desirability value (0.807). Conclusively, DOE helped in a complete understanding of the method, allowing for improved method development and subsequent method transfer with minimum systematic and inherent random variability. Based on the analysis of the Box-Behnken design, the optimal parameters were determined and tabulated in Table 8 for the developed method. Three sets of experimental runs were conducted as per optimized analytical conditions. The observed experimental results matched the model-predicted results, as tabulated in Table 9. The table provides a side-by-side view of predicted outcomes against actual observations, assessed at a two-sided 95% confidence interval (CI) and a 99% population interval.
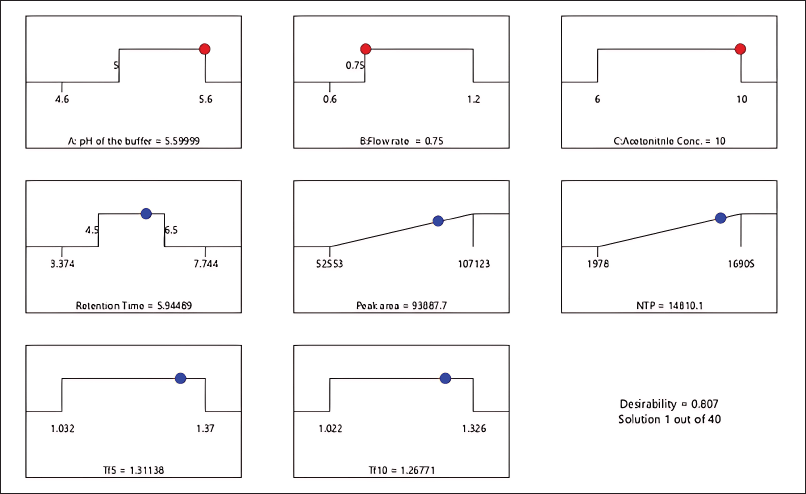 | Figure 4. Numerical optimization indicates an optimized solution with the highest desirability value. [Click here to view] |
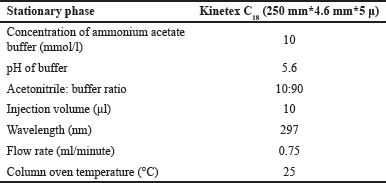 | Table 8. Optimal chromatographic conditions. [Click here to view] |
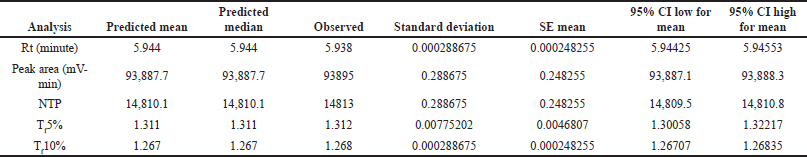 | Table 9. Predicted and actual point prediction at two-sided 95% CI. [Click here to view] |
Validation of the optimized HPLC method
The optimized HPLC method was validated by performing a rigorous set of experiments recommended by ICH Q2(R1) guidelines that assessed the method’s performance under various conditions. The results of all the validation parameters are listed in Table 10.
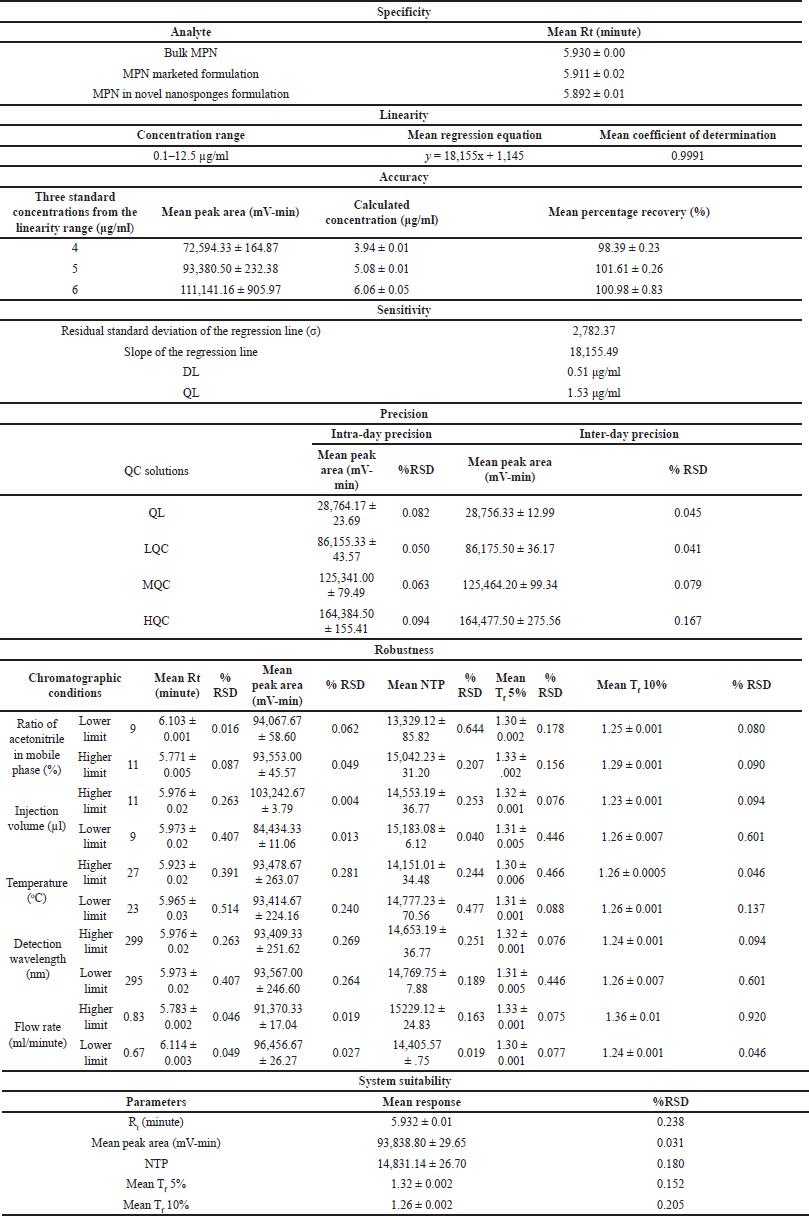 | Table 10. Results of all the validation parameters. [Click here to view] |
Specificity
The method represented a distinctive peak for MPN at a Rt of 5.93 minutes, as illustrated in Figure 5. No interferences were observed at the Rt of the MPN from the blank and placebo of the novel formulation. The developed method eluted the MPN from the marketed formulation and the novel nanosponges formulation at a Rt of 5.91 and 5.89 minutes, respectively. Hence, the optimized method was demonstrated to be highly specific in determining MPN.
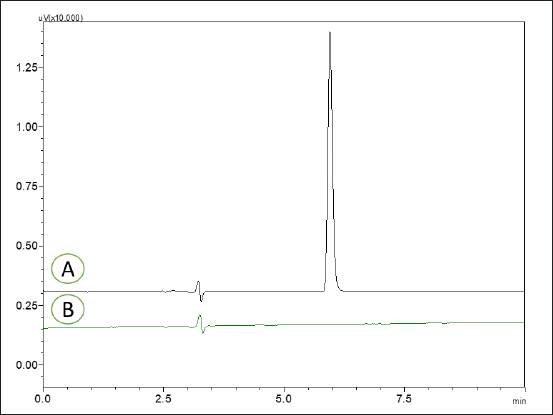 | Figure 5. HPLC chromatograms of MPN reference standard 5 μg/ml (A) and HPLC chromatograms of blank (B). [Click here to view] |
Linearity
MPN were analyzed in quintuplicate for a concentration range of 0.1 to 12.5 μg/ml. Individual linear plots were constructed for each trial, and the mean of all trials was calculated. According to the plotted linear regression graph, the linear regression equation for the mean value was y = 18155x+1145. The coefficient of determination (R2) was found to be 0.9991. Out of five linear regression plots, no two consecutive R2 values were less than 0.999. Therefore, the optimized method was significantly linear in the MPN concentration range and subjected to further experimentation.
Accuracy and precision
Three different MPN concentrations were injected in sextuplicate, and the mean percentage recovery of the concentrations 4, 5, and 6 μg/ml were 98.39%, 101.61%, and 100.98%, respectively. The accuracy results showed that the mean recovery percentage of analysis of MPN was well within the range of 85%–115%. Thus, the optimized method demonstrates suitability for the accurate quantification of MPN.
Four QC concentrations of MPN were injected in duplicate, and in the intraday analysis, the mean peak areas of the concentrations of 1.53, 4.59, 8.75, and 6.67 μg/ml were 28,764.17, 86,155.33, 125,341.00, and 164,384.5 mV-min, respectively. In the inter-day analysis, the same four QC concentrations of MPN were injected in duplicate; the mean peak areas of the concentrations of 1.53, 4.59, 8.75, and 6.67 μg/ml were 28,756.33, 86,175.5, 125,464.2, and 164,477.5 mV-min, respectively. The precision results showed that the % RSD of the peak area for all the concentrations in intraday and interday analyses was less than 2%. Thus, the optimized method demonstrates suitability for the precise quantification of MPN. The optimized technique has exhibited a high degree of suitability for the precise quantification of MPN.
The analytical performance of our developed method was statistically compared with the previously reported method by Mendez et al. [11] The precision comparison using F-test revealed that our method demonstrates significantly better precision, with %RSD values ranging from 0.041% to 0.167% compared to their reported values of 0.78% (intra-day) and 0.85% (inter-day). The calculated F-values were lower than the critical F-value at a 95% confidence level, confirming the superior precision of our method. In terms of accuracy, our method showed recovery values ranging from 98.39% to 101.61%, which was comparable to their reported range of 99.11%–100.10%. The Student’s t-test performed on the recovery data showed no significant difference between the two methods at a 95% confidence level, indicating that both methods are equally accurate. These statistical comparisons demonstrate that our method maintains equivalent accuracy while offering improved precision compared to the previously reported method.
Sensitivity
The DL was calculated based on the residual standard deviation of the regression line and its slope. The DL and QL were determined and found to be 0.51 and 1.53 μg/ml, respectively.
Robustness
An MPN concentration of 5 μg/ml was injected in triplicate, and the impact of small variations in the operating conditions, like the acetonitrile ratio in the mobile phase, column oven temperature, wavelength, flow rate, and injection volume, was analyzed. For all the varied conditions, the mean %RSD of peak area, Rt, number of theoretical plates, tailing factor 5%, and tailing factor 10% were less than 2. The optimized methodology has been demonstrated to have a high degree of robustness in obtaining precise and accurate quantifications of MPN in response to variations in operating conditions.
System suitability
An MPN concentration of 5 μg/ml was injected in sextuplicate and analyzed for mean Rt, peak area, number of theoretical plates, tailing factor 5%, and tailing factor 10%. The consistent results from sextuplicate trials ensure the suitability of the analytical system for the intended application of quantification of MPN in dosage forms.
Benchtop stability studies of MPN solution
The benchtop stability study was carried out to assess the stability of MPN in solution under ambient temperature conditions, and the results are depicted in Table 11. The analysis of lower and higher QC MPN concentrations (4.59 and 8.75 μg/ml, respectively) over time revealed that the stability of MPN was significantly affected by storage duration at ambient temperature. Results showed that the Rt of MPN remained the same (5.942 minutes), and the MPN concentration of 4.59 and 8.75 μg/ml decreased to 91.31% and 92.51% of the initial concentration after 24 hours, respectively, and to 77.38% and 78.91% after 48 hours, respectively. This implies that the MPN undergoes hydrolysis, which gradually converts the active form of the drug into the inactive form. The similarity index for the 48 and 24 hours stability was determined and tabulated in Table 11.
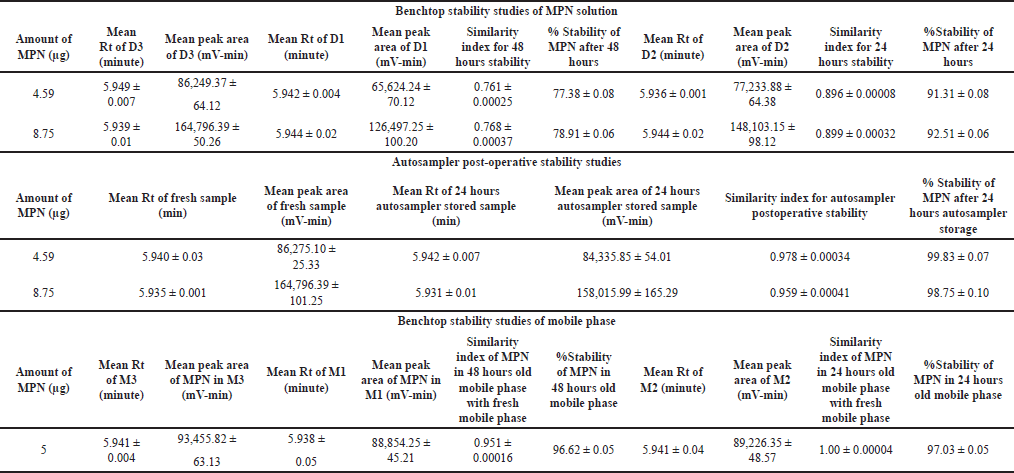 | Table 11. Results of stability studies. [Click here to view] |
Autosampler post-operative stability studies of MPN solution
Autosampler post-operative stability studies were conducted to assess the stability of MPN in solution form, stored in an autosampler at 4°C for 24 hours. Over time, the analysis of lower and higher QC MPN concentrations (4.59 and 8.75 μg/ml) revealed that storage duration at 4oC negligibly influenced the MPN stability. The results indicated that the Rt of MPN remained the same (5.937 minutes), and the MPN concentration of 4.59 and 8.75 μg/ml was stable and remained at 99.83% and 98.75% of the initial concentration after 24 hours at 4°C, respectively, as represented in Table 11. This implies that the rate of hydrolysis was significantly reduced at lower temperatures (refrigerated conditions).
Benchtop stability studies of the mobile phase
Benchtop stability studies were performed on the mobile phase to evaluate its stability at ambient temperature over a specific period and its ability to elute MPN. The analysis of the MPN concentration of 5 μg/ml over time showed that the stability of the mobile phase had a minimal impact on the concentration of MPN, and the Rt of MPN remained the same (5.940 minutes). The results indicated that the MPN concentration decreased to 96.62% and 97.03% of the initial concentration after 48 and 24 hours, respectively, as represented in Table 11. This demonstrates that the mobile phase was stable for 48 hours and can elute MPN effectively even after 48 hours.
Acid—induced degradation studies
Real-Time Conditions: MPN was subjected to 0.1 N and 1 N HCl at ambient temperature for 24 hours to evaluate its stability under acidic conditions. The analysis showed significant degradation, with MPN concentrations reducing by 82.23% and 89.16%, respectively. Degradation peaks were observed near the solvent front (1–3 minutes), indicating acid-catalyzed protonation of the MPN molecule. This reaction heightened molecular reactivity, leading to hydrolysis and the formation of inactive degradation products.
Accelerated Conditions: MPN solutions were exposed to 0.1 N and 1 N HCl at 60°C to simulate accelerated degradation. Substantial degradation was noted, with the primary peak showing splitting and additional peaks near the solvent front. The degradation process was attributed to combined acid-catalyzed hydrolysis and thermal effects, emphasizing the need for controlled storage conditions to maintain drug stability.
Alkali—induced degradation studies
Real-Time Conditions: Stability studies involved exposing MPN to 0.1 N and 1 N NaOH at ambient temperature. After 24 hours, degradation levels reached 75.09% and 91.19%, respectively. The basic environment deprotonated the MPN molecule, promoting beta-lactam ring cleavage and subsequent degradation.
Accelerated Conditions: MPN solutions in 0.1 N and 1 N NaOH were heated to 60°C. Significant degradation was observed, characterized by the splitting of the main peak and additional degradation peaks near the solvent front. The combined effects of base-catalyzed hydrolysis and thermal degradation led to the formation of less active products, indicating the susceptibility of MPN to alkaline and high-temperature conditions.
Oxidative degradation studies
Real-Time Conditions: MPN stability was evaluated by exposing solutions to 3% hydrogen peroxide (H2O2) at ambient temperature for 24 hours. Results indicated 30% degradation, likely due to oxidation of the sulfur atom in the thiazolidine ring or cleavage of the beta-lactam ring.
Accelerated Conditions: To simulate oxidative stress, MPN solutions were treated with 3% H2O2 and heated to 60°C. Accelerated degradation was evident, with peak splitting and new degradation peaks near the solvent front. Free radical formation and elevated temperatures exacerbated bond cleavage and molecular destabilization.
Photolytic degradation studies
Photolytic stability was assessed by exposing MPN to UV light and direct sunlight for 24 hours. The concentration of MPN decreased by 20%, driven by photodegradation. UV-induced bond cleavage formed unstable degradation products, with peaks observed near the solvent front. This study underscores MPN’s sensitivity to light exposure and highlights the importance of protecting the drug from UV and sunlight.
Application of the developed analytical method for quantification of MPN in marketed products and novel nanoformulations
The validated HPLC method was successfully applied for the quantification of MPN in a marketed powder for injection formulation (Merosure 125 mg, Alkem Laboratories, India) and a novel β-CD nanosponges formulation, demonstrating its feasibility for real-world applications.
Quantification of MPN in the marketed formulation
A stock solution of 10 μg/ml of MPN prepared from the marketed product was analyzed using the developed method. The results showed consistent Rt and peak area, with a recovery rate of 99%, highlighting the method’s high accuracy and precision.
Quantification of MPN in β-CD-nanosponges
A stock solution of 10 μg/ml of MPN prepared from the marketed product was analyzed using the developed method. The results showed consistent Rt and peak area, with a recovery rate of 99%, highlighting the method’s high accuracy and precision.