
INTRODUCTION
Millions globally are living with HIV, with access to antiretroviral therapy (ART) transforming the disease into a manageable condition [1]. However, adherence to ART remains crucial, as lapses can lead to treatment failure and drug resistance [2]. This poses a significant challenge for patients with multidrug-resistant HIV-1, increasing their risk of complications and death [3]. Lenacapavir (LCV) emerges as a groundbreaking treatment option in this scenario [4]. It belongs to a new class of HIV-1 drugs known as capsid inhibitors [5]. By targeting the viral capsid, LCV disrupts HIV replication at multiple stages [6]. Clinical trials have shown promising results, with a significant reduction in viral load compared to placebo [7]. This effectiveness led to its approval by the FDA in December 2022 for treating HIV-experienced patients [8]. LCV offers a distinct advantage with its slow-release properties. This allows for flexible dosing options, including daily or weekly oral administration and a remarkable extended dosing interval of every 6 months via subcutaneous injection. This infrequent dosing is attributed to LCV’s extended half-life, particularly in its injectable form. Additionally, LCV undergoes minimal metabolism, further contributing to its sustained presence in the body [9]. However, a crucial aspect to consider is the impact of certain medications on LCV’s effectiveness. Rifampicin, a strong inducer of a specific metabolic pathway, significantly reduces LCV exposure in plasma. Due to this interaction, co-administration of these drugs is contraindicated [10]. Given the complexities associated with multidrug-resistant HIV-1 infection, ensuring both safety and efficacy is paramount for clinicians. Monitoring LCV plasma concentrations, similar to other long-acting anti-retrovirals, may be necessary to guarantee consistent and adequate drug exposure throughout the extended dosing interval. LCV quantification currently lacks robust options. Only two analytical methods exist [11] underscoring the need for more effective and sensitive approaches. This research contributes to this goal by describing the development and validation of a new analytical method for quantifying LCV levels in rat plasma using high-performance liquid chromatography-tandem mass spectrometry. Figure 1 shows the Structure of LCV.
 | Figure 1. Chemical structure of LCV. [Click here to view] |
MATERIALS AND METHODS
Chemicals and reagents
The reference sample was provided as LCV samples from Glenmark Life Sciences Ltd, Mumbai. LCMS grade Acetonitrile, LCMS grade Methanol and all other chemicals were obtained from Merck chemical division, Mumbai. HPLC-grade water obtained from Milli-Q water purification system was used throughout the study.
LC-MS/MS instrument and conditions
Chromatography was performed with waters 2,695 HPLC provided with a high-speed auto sampler, column oven, and degasser and SCIEX QTRAP 5,500 mass spectrometer to provide a compact and with class Empower-2 software.
The mass spectrometer was managed in positive ion electrospray ionization interface mode. Multiple reactions monitoring mode has been applied to quantify the LCV. Working parameters have been set as follows: Collision energy: 14 V; Ion spray voltage: 5,500 V; Source temperature: 550°C; Drying gas temperature: 120°C–250°C; Collision gas: nitrogen; Drying gas flow stream: 5 ml/minute; Declustering potential: 40 V; Entrance potential: 10 V; Exit Potential: 7 V; Dwell time: 1 second; Curtain gas (CUR): 12 psi; Collision gas (CAD): 10 psi. LCV and IS were identified using proton adducts in the LC-MS analysis at m/z 969.32/509.15 and 975.28/515.07, respectively. Figure 2 shows the MRM of (A) LCV(B) LCV-IS in Positive Polarity: Analogous Precursor Ion and Daughter Ion with the Highest Intensity (m/z).
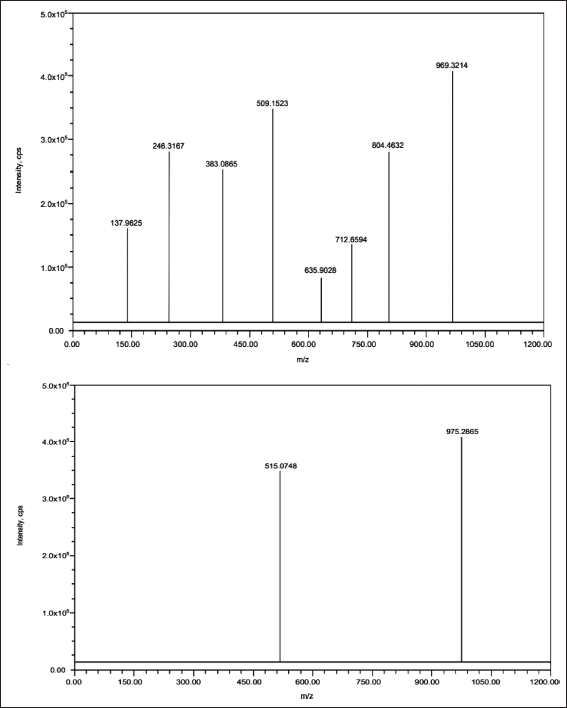 | Figure 2. MRM of (A) LCV(B) LCV-IS in positive polarity: analogous precursor Ion and Daughter Ion with the highest intensity (m/z). [Click here to view] |
Chromatographic conditions
For the chromatographic analysis, a waters Symmetry C18 column, 150 × 4.6 mm with a particle size of 3.5 µm was selected. The mobile phase was a combination of acetonitrile and a buffer containing 0.1% formic acid, and it was given at a flow rate of 1 ml per minute. The ratio of the two components was 20:80. Throughout the entirety of the analysis, the temperature of the column did not deviate from the standard setting. LCV-IS was chosen as the internal standard due to its suitability in terms of chromatographic performance and extractability. Each sample was injected into a volume of 10 µl. During chromatographic analysis, the retention time for LCV& LCV-IS was approximately 2.715 minutes, while the run time was 5 minutes.
Development of calibration standards and quality control samples
Standard stock solutions of LCV and LCV-IS were formulated at a concentration of 200 ngml−1. A working standard solution of 50 ngml−1 was produced by suitably diluting the LCV master stock solution, originally at 200 ngml−1, using the mobile phase. The LCV standard solutions that are suitably diluted from the master stock solution were then added to drug-free rat plasma. This resulted in LCV concentrations of 5, 12.5, 25.00, 37.5, 50, 62.5, 75, and 100 ngml−1 for the analytical calibration standards. In addition to calibration standards, QC standards were established with specific LCV concentrations of 5, 25, 50, and 75 ngml−1. The purpose of these quality control samples is to track the development and dependability of the analytical method. Every calibrated and quality control standard that was prepared was preserved in a freezer set to −30°C.
Sample preparation for LCV and LCV-IS from rat plasma
Liquid–liquid extraction was employed for the isolation of LCV and its respective internal standard LCV-IS from rat plasma samples. To begin, a volume of 500 μl of diluent was added to 200 μl of plasma, ensuring that the mixture was thoroughly combined. Following this, 300 μl of acetonitrile was introduced to precipitate all proteins, and the mixture was vigorously vortexed. Subsequently, the mixture was centrifuged at 4,000 RPM for a duration of 15–20 minutes, resulting in the separation of the supernatant solution. For the preparation of the samples for analysis, the supernatant from each sample was meticulously transferred into appropriately labeled RIA vials. Subsequently, the solvent underwent evaporation at 40°C until complete desiccation was achieved. Following this, the desiccated samples were reconstituted with 500 μl of the mobile phase, undergoing a brief, vigorous mixing process for homogeneity. Finally, these reconstituted samples were transferred into auto sampler vials, rendering them ready for injection into the chromatograph.
Method validation
Linearity
This study exhibits the linearity of detector response by utilizing eight different concentrations of an LCV solution, ranging from 5 to 100 ngml−1. The range of concentrations was chosen to demonstrate the linearity of the detector response. The experiment utilized a method of extraction that was consistent with the goal of validating the linearity of the detector’s response while assessing varied concentrations of LCV solutions that were put into rat plasma. As the Internal Standard, we utilized a solution with a constant concentration of 50 ngml−1. The HPLC system was then fed with the sample concentrations that had been determined as a consequence. The data that was collected, which represented the ratio of LCV peak area to LCV-IS peak area, was used to generate a correlation plot relating the ratio with LCV concentration in ngml−1. The correlation plot was linked to the ratio. The regression analysis was used to determine important parameters such as the correlation coefficient, slope, intercept, LOD, and LOQ.
Selectivity and specificity
By corresponding their distinct retention times to the corresponding MRM responses, the chromatographic peaks of LCV and LCV-IS were discerned. A criterion was established to evaluate selectivity: the peak area of LCV at its specified retention time in blank samples must not surpass 20% of the mean peak area of the LCV’s Limit of LOQ. The mean peak area of the IS’s LOQ should not exceed 5% of the peak area at its retention time in blank samples.
Precision and accuracy
Intraday and interday precision were determined through a rigorous evaluation of six replicates of each concentration at the LQC level (25), LLOQ level (5), MQC level (50), and HQC level (75). The evaluation of inter-day precision spanned multiple days, whereas intra-day precision was determined by analyzing these replicates on the same day. To determine the coefficient of variation (RSD), comparisons were made between the measured and expected true responses at the QC levels using the resulting area response ratio values. A criterion of 20% accuracy was established for the LLOQ level, signifying that the measured values ought not to exhibit a greater than 20% deviation from the true values. A 15% acceptable accuracy criterion was subsequently established for the HQC, MQC, and LQC levels.
%Recovery
In the assessment of LCV and LCV-IS extraction recovery from rat plasma, a rigorous procedure was conducted, involving the analysis of six replicate injections of quality control samples. These QC samples included LQC, MQC, and HQC with corresponding concentrations of 25, 50, and 75 ngml−1. This determination was made by comparing the peak areas derived from the extracted plasma samples with the peak areas from a standard solution that had been spiked with the blank plasma residue. It is imperative to note that a recovery rate exceeding 50% was the established criterion for adequacy in achieving the requisite sensitivity in this analytical process.
Matrix effect
To evaluate the impact of the matrix, six batches of empty biological matrices were created. Each batch was then mixed with the pure standard at two different concentration levels, specifically the LQC and HQC levels, with each concentration level being tested three times. Subsequently, the spiked samples were compared to the neat standards of identical concentration by means of alternate injections. To determine the dependability of the matrix factor, total accuracy was used as a primary measure. The level of precision was quantified as CV%. The acceptable threshold for CV% was established at 15% or below, which is worth mentioning.
Stability
The experimental design comprised six repetitions for each of the three concentration levels (LQC, MQC, and HQC). In accordance with FDA regulations, the stability of an analyte is assessed by monitoring the extent of variation in its concentration within a 15% threshold. To evaluate resistance to repeated freezing and thawing, freeze-thaw stability was determined by subjecting samples to three consecutive defrost cycles at −31°C, followed by a comparison with freshly injected internal control samples after the third cycle. The bench-top stability study involved storing QC samples at these concentrations at room temperature for 24 hours. Subsequently, these samples were compared to plasma extracts that were immediately analyzed. To ensure short-term stability, six replicates of LQC, MQC, and HQC samples were maintained at 7°C for 7 days. After examining the long-term stability for 1, 7, 14, 21, and 28 days, samples were analyzed. To evaluate the analyte’s stability under prolonged exposure, the auto-sampler stability test utilized three samples of the specified QC levels that were stored in an auto-sampler at 15°C for durations spanning from 0 to 24 hours. In the dry and moist extract plasma sample evaluation, the stability of samples stored at ambient temperature for specified times—12 and 18 hours—within the range of 2°C–8°C was compared. The evaluation encompassed a comparison with newly extracted samples to determine the stability of the analyte by ensuring that the RSD remained below 15%.
Application to the pharmacokinetic study of LCV in rat plasma
The liquid–liquid extraction method was used to isolate LCV in rat plasma. For this, 200 µl of plasma sample (respective concentration) were added into labeled polypropylene tubes and vortexed briefly after that 500 µl of standard stock and 500 µl of Internal standard stock were added and vortexed for approximately 10 minutes followed by centrifuge at 4,000 rpm at 20°C. Supernatant from each sample was transferred to labeled via tube and evaporated at 40°C until dryness. This sample was reconstituted with 300 µl of methanol and 500 µl of diluents vortexed briefly and then transferred the sample into auto sampler vials for injection.
LCV sample was injected into rat bodies and collected samples at different time intervals such as 1, 2, 3, 4, 5, 10, 24, 48, 120, and 240 hours in six different rats. After that samples are prepared as per the test method and injected into a chromatographic system to record their values. A single dose of LCV tablets (300 mg) was administered rats, samples were collected at 1, 2, 3, 4, 5, 10, 24, 48, 120, and 240 hours post-dose. An aliquot of 300 µl blood was collected at each time point in K2 EDTA vacutainer tubes. Additionally, a predose sample was collected to check the possible interferences from the plasma. The collected samples were centrifuged to obtain the plasma and stored at −70°C. Plasma samples were spiked with the IS and processed along with QC samples at four concentrations. The pharmacokinetic parameters of LCV were calculated using WinNonlin (Version 5.2) software package. The stability of the study samples was established by incurred sample reanalysis (ISR). For ISR, two samples from each subject were selected near Cmax and the elimination phase in the pharmacokinetic profile. The samples were considered stable; the percent difference should not be more than 20%.
RESULTS
Method validation
Linearity
The evaluation of linearity was conducted by creating calibration curves plotting the ARR (LCV/IS) against LCV concentration. The calibration demonstrated linear behavior over a concentration range from 5 to 100 ngml−1. Statistical analysis of the data resulted in a high correlation coefficient (r2) value of 0.9999. Additionally, the determined intercept and slope were found to be 0.025406 and 0.0201, respectively. Refer to Figure 3 for the visualization of the calibration plot.
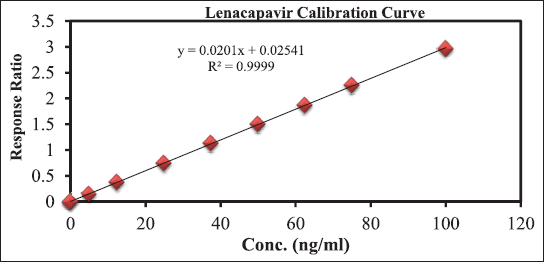 | Figure 3. For the visualisation of the calibration plot. [Click here to view] |
The utilization of the MRM function to analyze LCV and IS yielded extremely selective results, which was confirmed by the absence of interference in the chromatograms of rat plasma at retention times of LCV and IS. The chromatograms of the blank and blank spiked with LLOQ, LQC, MQC, and HQC samples along with ISD are displayed in Figure 4.
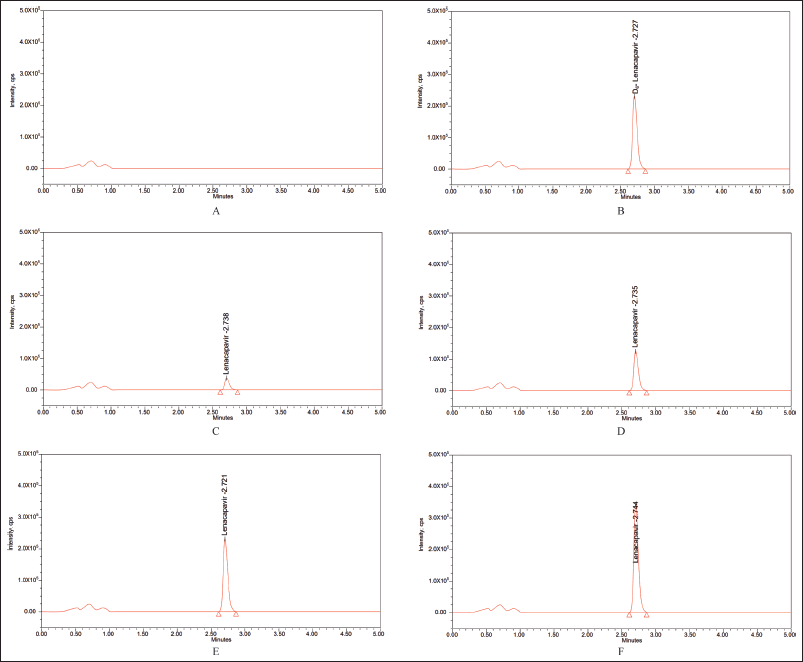 | Figure 4. Blank plasma chromatogram (A) devoid of any interferences; representative chromatograms for (B) rat plasma samples spiked with IS, (C) LLOQ = 5, (D) LQC = 25, (E) MQC = 50, and (F) HQC = 75. [Click here to view] |
Accuracy and precision
In the evaluation of intraday accuracy, the accuracy percentages for the specified concentrations across the four concentration checkpoints of LLQC, LQC, MQC, and HQC were determined to be 90.0, 98.77, 100.14, and 100.96. Similarly, in the inter-day precision study, the accuracy percentages for these concentrations were consistently observed to be 100.1, 100.06, 100.01, and 100.02, as shown in Table 1.
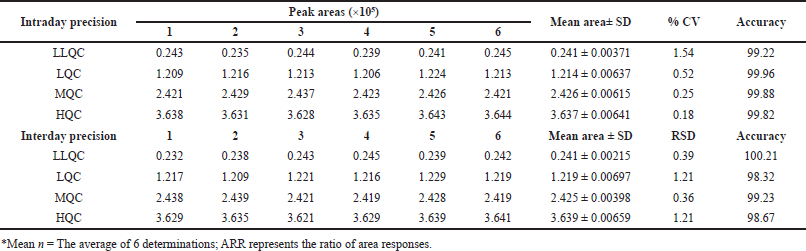 | Table 1. Precision and accuracy evaluation of PC. [Click here to view] |
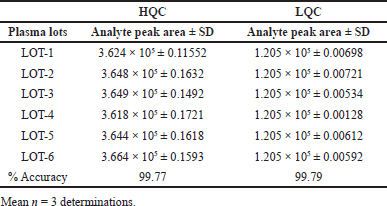
Recovery
Figuring out LCV recovery involved directly comparing the variance in peak area ratios between plasma and solvent samples. This assessment was conducted at three distinct concentrations: 25, 50, and 75, yielding recovery percentages of 100, 99.7, and 99.8, respectively. It is noteworthy that the % CV remained well within the acceptable limits.
Matrix interference
The mean percent accuracy for the matrix effect was calculated to be 99.9% and 98.4%, respectively, which falls within the permissible range of 80%–120%. The percent SD of the substance at both the LQC and HQC levels was acceptable. As summarized in Table 2, this indicates that the effect of the matrix on the ionization of the analyte is within the acceptable range.
 | Table 2. Matrix effects of PC. [Click here to view] |
Stability
The precision of LCV sample accuracy was rigorously ascertained via an exhaustive bench-top stability investigation; the resulting values of 97.33, 99.48, and 98.56 correspond to distinct levels. Additionally, at the LQC, MQC, and HQC levels, the accuracy pertaining to freeze-thaw stability was assessed; the corresponding accuracy values were 100.87, 99.22, and 100.18. The results obtained from assessing the samples’ stability in the short and long term, as indicated by the RSD remaining within 15% of the predetermined acceptability threshold, demonstrate that these samples maintained their stability for a maximum of 28 days. The consistency of auto-sampler outcomes, which include values of 98.3, 98.5, and 99.7, highlights the superior stability exhibited by processed samples after undergoing auto-sampling in comparison to freshly prepared samples. The information presented in Table 3 illustrates the overall stability test results, which indicate that the LCV samples remain within the permissible range of variation throughout the entire analysis procedure.
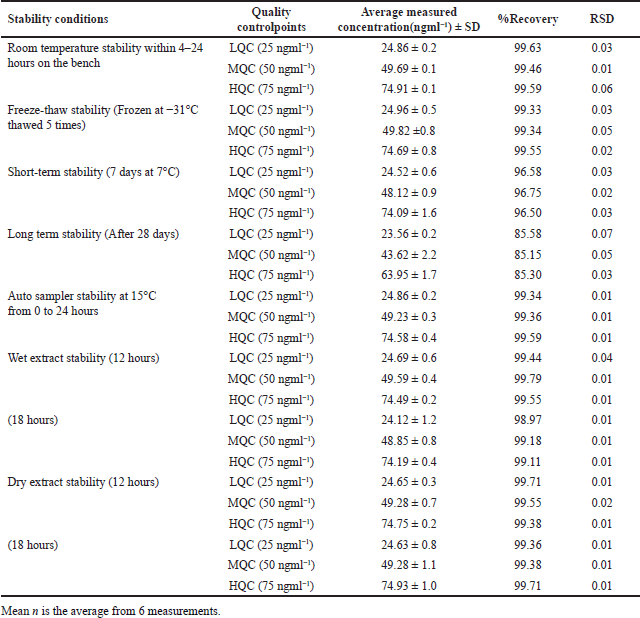 | Table 3. LCV Stability findings in rat plasma. [Click here to view] |
Pharmacokinetic investigations
A formal investigation of pharmacokinetic parameters was conducted following the administration of a sole LCV injection dose in six rodents, employing a non-compartmental timing analysis approach. Plasma concentrations of LCV were meticulously measured at specified intervals of 5, 10, 20, 30, 40, 50, and 60 m post-dosage, and the mean of these collective concentration-time profiles are visually depicted in Figure 5. The outcomes of this investigation yielded crucial pharmacokinetic parameters: Cmax of 48.415 ± 0.28, Tmax at 4 ± 0.002, T1/2 of 120 ±0.01, AUC0-t at 9851 ± 5.4 ng h ml-¹, and AUC0-∞ at 9851 ± 5.4 ng h ml. A comprehensive summary in Table 4 underscores the method’s effectiveness for bioanalytical investigations while providing valuable data for preclinical and pharmacokinetic research endeavors. The protocol of the animal study was approved by the Institute of Animal Ethics Committee (Reg.No:1250/PO/RcBi/S/19/CPCSEA).
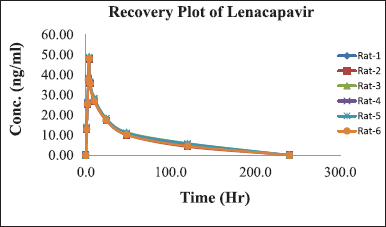 | Figure 5. Graph depicting the time course of mean plasma concentrations of LCV following intravenous administration in rats. [Click here to view] |
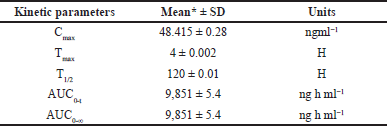 | Table 4. Mean pharmacokinetic parameter values of LCV following intravenous administration in rats with standard deviation. [Click here to view] |
DISCUSSIONS
In the quest for efficient analysis, the optimization of LC-MS conditions assumes importance, which involves a series of deliberate trials to fine-tune chromatographic parameters, particularly those pertaining to the mobile and stationary phases. The initial endeavors, employing an inertsil C18 column, 150 × 4.6 mm, 3.5 µ, when paired with a mobile solvent comprising 70:30 ratio of acetonitrile to formic acid buffer, resulted in plate count values deviating from the intended limits. Subsequently, when the mobile phase composition was altered to a 60:40 ratio, the first chromatographic peak exhibited an unfortunate tendency to split, further complicating the quest for optimal separation. Not giving up, the experiments went on, this time using a Thermosil C18 column, 150 × 4.6 mm, and a mobile phase of 80:20 Acetonitrile to Formic Acid Buffer. This arrangement introduced an unknown peak that emerged. Meanwhile, shifts in the mobile phase composition, specifically Acetonitrile and Formic Acid Buffer at ratios of 60:40 and 50:50, revealed inadequacies in base line quality and inter-peak resolution, respectively. The turning point in this optimization journey arrived with the adoption of an eluent mixture at a 20:80 ratio of acetonitrile and a 0.1% formic acid buffer. Under these conditions, utilizing the Waters Symmetry C18 column, 150 × 4.6 mm; 5 µ, an exquisite balance between separation and elution was achieved. With a judicious rate of flow of 1 ml/minute and a sample volume of 10 µl, the chromatographic system delivered the desired outcome. In parallel with chromatographic refinements, mass spectrometry optimization was diligently executed. By directly infusing solutions of both LCV and IS into the ESI source, meticulous adjustments were made to parameters such as nebulizers and desolvation gases. These refinements aimed to secure an optimal spray shape, fostering superior ionization and droplet drying. For LCV precursor ions, MH is cantered at m/z 969.32 with a fragment ion selected at m/z 509.15. In the case of IS, the precursor ion MH+ at m/z 975.28 was meticulously observed, with the fragment ion registering at m/z 515.07. Linearity results demonstrated a direct and proportional relationship. The recovery results clearly show that the bioanalytical method works to get a high extraction rate, which supports its suitability for strong and accurate quantitative analysis. The consistently high accuracy percentages at various QC levels demonstrate the method’s ability to produce reliable and consistent results. In rigorous stability assessments, the concentrations of PC samples were observed to exhibit a variation of no more than 15% in comparison to fresh samples.
CONCLUSION
The developed method has exhibited remarkable selectivity, and linearity, along with ruggedness and reproducibility. Employing a simple liquid–liquid extraction technique with minimal matrix interference and short retention times of less than 5 minutes, this method ensures a relatively swift analysis process. Noteworthy are the exceptional recovery rates, reaching nearly 100% at both high and low concentrations, and the extensive stability of LCV in rat plasma. The precision within and between batches (%CV) at (LLOQ, LQC, MQC, and HQC) levels falls below 15%. Notably, this bioanalytical approach, utilizing LC-MS/MS, provides the first-ever evaluation of the pharmacokinetics of LCV and holds the potential to greatly facilitate the pharmacokinetic assessment of LCV in rats, a crucial step in elucidating its safety, toxicity, and efficacy profiles, particularly in the context of anticancer research.
ACKNOWLEDGMENTS
The authors express their heartfelt appreciation to the administration of Dr. Samuel George Institute of Pharmaceutical Sciences, ANU University, Guntur, for granting them access to the facilities that greatly supported their research efforts.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to the conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
FINANCIAL SUPPORT
There is no funding to report.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
The protocol for the animal study was approved by the Institute of Animal Ethics Committee (Reg. No: 1250/PO/RcBi/S/19/CPCSEA).
DATA AVAILABILITY
All data generated and analyzed are included in this research article.
PUBLISHER’S NOTE
All claims expressed in this article are solely those of the authors and do not necessarily represent those of the publisher, the editors and the reviewers. This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
USE OF ARTIFICIAL INTELLIGENCE (AI)-ASSISTED TECHNOLOGY
The authors declares that they have not used artificial intelligence (AI)-tools for writing and editing of the manuscript, and no images were manipulated using AI.
REFERENCES
1. Teeraananchai S, Kerr SJ, Amin J, Ruxrungtham K, Law MG. Life expectancy of HIV-positive people after starting combination antiretroviral therapy: a meta-analysis. HIV Med. 2017 Apr;18(4):256–66. CrossRef
2. Cohen MS, Chen YQ, McCauley M, Gamble T, Hosseinipour MC, Kumarasamy N, et al. Prevention of HIV-1 infection with early antiretroviral therapy. N Engl J Med. 2011 Aug 11;365(6):493–505. CrossRef
3. Tolley EE, Li S, Zangeneh SZ, Atujuna M, Musara P, Justman J, et al. Acceptability of a long-acting injectable HIV prevention product among US and African women: findings from a phase 2 clinical Trial (HPTN 076). J Int AIDS Soc. 2019 Oct;22(10):e25408. CrossRef
4. Cobb DA, Smith NA, Edagwa BJ, McMillan JM. Long-acting approaches for delivery of antiretroviral drugs for prevention and treatment of HIV: a review of recent research. Expert Opin Drug Deliv. 2020;17(9):1227–38. CrossRef
5. Dvory-Sobol H, Shaik N, Callebaut C, Rhee MS. Lenacapavir: a first-in-class HIV-1 capsid inhibitor. Curr Opin HIV AIDS. 2022 Jan 1;17(1):15–21. CrossRef
6. Singh K, Gallazzi F, Hill KJ, Burke DH, Lange MJ, Quinn TP, et al. GS-CA compounds: first-in-class HIV-1 capsid inhibitors covering multiple grounds. Front Microbiol. 2019 Jun 20;10:1227. CrossRef
7. Lee NE, Sutherland RK. Lenacapavir and the novel HIV-1 capsid inhibitors: an emerging therapy in the management of multidrug-resistant HIV-1 virus. Curr Opin Infect Dis. 2023 Feb 1;36(1):15–9. CrossRef
8. Link JO, Rhee MS, Tse WC, Zheng J, Somoza JR, Rowe W, et al. Clinical targeting of HIV capsid protein with a long-acting small molecule. Nature. 2020 Aug;584(7822):614–8. CrossRef
9. Segal-Maurer S, DeJesus E, Stellbrink HJ, Castagna A, Richmond GJ, Sinclair GI, et al. Capsid inhibition with Lenacapavir in multidrug-resistant HIV-1 infection. N Engl J Med. 2022 May 12;386(19):1793–803. CrossRef
10. Temereanca A, Ruta S. Strategies to overcome HIV drug resistance-current and future perspectives. Front Microbiol. 2023 Feb 16;14:1133407. CrossRef
11. West RE 3rd, Oberly PJ, Riddler SA, Nolin TD, Devanathan AS. Development and validation of an ultra-high performance liquid chromatography-tandem mass spectrometry method for quantifying lenacapavir plasma concentrations: application to therapeutic monitoring. J Chromatogr B Analyt Technol Biomed Life Sci. 2023 Oct 15;1230:123905. CrossRef