
INTRODUCTION
Mild cognitive impairment (MCI) is defined as an intermediate stage between normal cognitive aging and pathological cognitive decline, such as major neurocognitive disorders (MNDs), including Alzheimer’s disease [1,2]. The pathophysiological process must be understood as a multifactorial process that includes cellular and molecular dysfunctions [3]; furthermore, MCI shares some signaling pathway alterations with MND such as Alzheimer’s disease [3–6], in this sense, MND produces high morbidity and mortality rates in older individuals around the world and current treatments have low possibilities of changing the conclusion of MND; however, MCI is significantly likely to be treated.
Current pharmacological treatment against MCI includes: 1) acetylcholinesterase inhibitors, necessary to avoid symptoms of MCI without halting its progression to MND; 2) a promising treatment combining physical exercise, cognitive training, and diet, suggesting a reduction of symptomatic MCI [6]. In addition, phytochemicals have several biological effects on the central nervous system: they act as neuroprotectors in the processes of oxidative stress and apoptosis, immunomodulate, and neuroregulate nervous tissue, among many others. Additionally, a diet rich in phytochemical compounds has been shown to influence specific molecular markers in MCI and MND [6–10].
This study focused on discovering possible phytochemical treatments for MCI. We conducted a virtual screening (VS) process to investigate the interactions of 2,588 phytochemical compounds with five key proteins that are dysregulated in MCI. The selected markers used in this research are as follows:
1) β- Secretase (BACE1) is a protease involved in the cleavage of amyloid precursor protein (APP) [11–13]; subsequently, APP is cleaved by γ-secretase, producing amyloid β peptide (A-β), some studies indicate that an increase in β-secretase activity leads to an excess of A-β plaques, which contributes to neurotoxicity [14,15]. 2) Glycogen synthase 3-β (GSK3β) plays a significant role in Tau phosphorylation [16] and is involved in various physiological processes. However, when GSK3β is overexpressed, it causes an increase in Tau phosphorylation, which leads to the destabilization of microtubules [5]. 3) Glutamate transporter associated protein 3–18 (GTRAP3-18) blocks the translocation of the early excitatory amino acid carrier 1 in the endoplasmic reticulum. This interaction leads to an increase in reactive oxygen species and a reduction in glutathione, causing an imbalance in oxidative processes within neurons [17,18]. 4) Pregnane X receptor (PXR) is a regulator of P-glycoprotein, and its activation produces an increased clearance of A-β and reduction of MCI [19,20]. 5) Epidermal growth factor receptor (EGFR) in Brain injuries and pathological conditions like MCI and MND increase its expression and activation reducing Akt levels [21], which plays an inhibitory activity against GSK3β. On the other hand, EGFR could activate reactive astrocytes and increase inflammatory response in MCI and MND [22,23].
Following the VS analysis, we selected the best phytochemical candidates based on docking scores. Subsequently, we conducted docking analysis and ligand-receptor molecular dynamics (MDs) to determine the most probable conformation of the protein-ligand complex. Finally, the methods used in this study provide a set of phytochemical compounds with potential activity against MCI.
MATERIALS AND METHODS
3D ligand structures retrieval and optimization
A total of 2,568 phytochemical compounds from different libraries were used in this study. First, from PubChem database [24], through compound subclassification Kyoto Encyclopedia of Genes and Genomes (KEEG), we select alkaloids, flavonoids, and terpenes, we downloaded comma-separated values libraries with 714 alkaloids, 478 flavonoids, and 1,102 terpenoids; other 16 polyphenols were obtained from Phenol-explorer [25], 76 sulfur-containing compounds were taken from SuCComBase [26], 33 isocyanates from the base “chemical entities of biological interest” (ChEBI) [27] and 149 glucosinolates were taken from Blaževi? et al. [28] and drew in ZINC database [29]. Using OpenBabel [30], 3D conformers of the 2,568 phytochemical compounds were obtained from the simplified molecular input line entry system (SMILES) to 3D structures, minimized by 1,500 steps of conjugate gradient algorithm considering Merck Molecular force Field (MMFF94).
Protein preparation and molecular dynamics
The structure of EGFR, GSK3β, BACE1, and PXR were taken from RCSB Protein Data Bank [31], (PDB ID 1M17), (PDB ID 4J71), (PDB ID: 2ZHT), and (PDB ID: 3CTB), respectively; on the other hand, GTRAP3-18 structure was taken from AlphaFold [32, 33] (Alpha ID: Q9ES40), and subjected to 200 ns of (MDs) simulation to optimize the model and reach a stable conformer. The other four proteins were subjected to 100 ns of MDs after the removal of other molecules different from the main protein chain using USCF Chimera [34].
Simulations were conducted under the parameters of CHARMM36-jul2020 force field in GROMACS 2021.4 [35,36]. Using a cubic box model, filled with TIP3 water model based on a simple point charge, and 0.15 M of NaCl which also served to neutralize the system. The system was subjected to 50,000 steps of the steepest descent minimization process, with maximum force set to 10 KJ/mol. After that, an equilibration with 100 ps of a canonical ensemble (NVT) and an isothermal-isobaric ensemble (NPT) equilibration was performed.
Subsequently, the simulations were carried out in the NPT ensemble for 100 ns at 300 K and 1 atm, V-rescale and Parrinello-Rahman algorithms were used to sample canonical temperature and pressure conditions. A LINCS algorithm with a cut-off of 10 Å was used for computing long-range electrostatic interactions by the Particle Mesh Ewald method. In the same way, 10 Å was the limit to calculate non-bonded interactions, i.e., Coulomb (electrostatic) and Lennard Jones (hydrophobic/van der Waals attractions) using the Verlet cut-off scheme. The leap-frog algorithm was used to compute the equation of motion with a time step of 2 fs.
After completing the MDs simulations for each protein, the GROMACS clustering algorithm using a cutoff of 0.2 nm was applied. This allowed us to identify clusters present throughout the entire trajectory of the simulations.
Virtual screening
Based on clustering analysis, those clusters that represent at least 60% of each protein whole dynamics were obtained, using AutoDock Tools [37], hydrogens and Kollman charges were added, and the coordinates of a grid box centered on each protein catalytic site (based on co-crystallised agonist or antagonist), with a 20 × 20 × 20 Å size volume on each one were obtained; after these, each cluster (saved as pdbqt) was used as the target for VS using Vina [38,39]. VS was performed with a bash script. A single pose was analyzed based on protein-ligand interactions and binding affinity values to perform this process. The best ligand of each cluster (for each protein) was used to develop receptor-ligand MDs.
Molecular docking
Based on VS results, the best two ligands for each cluster were used to perform 1,000 independent replicates by using a local bash script to obtain the most representative ligand-protein conformation. Based on docking scores, interactions, and Root Mean Square Deviation calculation, the most probable ligand-protein conformer was chosen to carry out MDs.
Ligand-receptor MDs
MD of the best ligand-receptor conformation was performed in GROMACS 2021.4 using CHARMM36-Jul2020 forcefield [35,36], considering the same conditions as in point 4. We utilized each ligand, including those co-crystallized, to generate topologies using the CgenFF online server [40,41]. The average structure representing the whole MDs simulation was obtained after clustering analysis with a 0.2 nm cutoff, which was used to build representative images on PyMOL [42], and the 2D interaction images were obtained with Biovia Discovery Studio [43].
RESULTS AND DISCUSSION
MDs for clusters
We performed a clustering analysis To determine the most common and stable conformers in protein structures over a designated timeframe. This analysis allowed us to investigate whether the binding site of each protein varies under dynamic conditions, which in turn affects ligand binding, enabling us to provide a more accurate description of binding behavior. As illustrated in Figure 1, BACE1 and GSK3β exhibited a single cluster, depicted in orange, indicating that both proteins maintained minimal conformational changes throughout the 100 ns simulation, with movements remaining under 0.25 Å. The other three proteins (GTRAP3-18, PXR, and EGFR) each displayed three clusters. The most stable conformation is shown in orange, the second most stable in green, and the least common form in blue. Each cluster represents at least 10 ns of the total simulation, suggesting that each can be considered a viable conformation for different ligand binding modes.
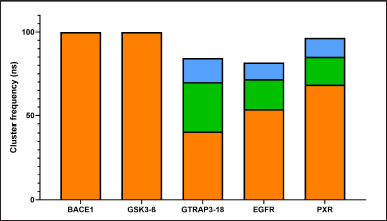 | Figure 1. Main clusters of the MDs of the analyzed proteins. Cut-off= 0.25 Å; ns = nanoseconds. The most stable conformation is shown in orange, the second most stable in green, and the least common form in blue. [Click here to view] |
Virtual screening
As a first approach, a VS of 2,568 phytochemical compounds with the five key proteins was performed to find the ten best compounds with the higher binding score. Figures 2 and 3 depict how most compounds binding to the five proteins are alkaloids, with a few terpenes and only three belonging to the phenolic compound family.
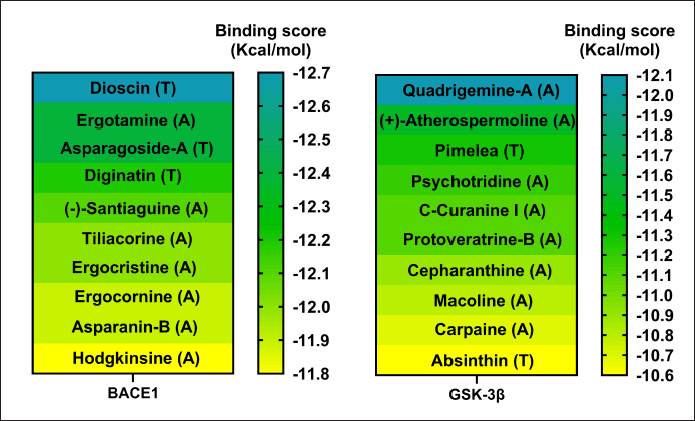 | Figure 2. Docking scores of the best 10 compounds were obtained after MDs clustering of BACE1 and GSK3β (Only a single cluster was found for these two proteins). Considering ligands as alkaloid (A), terpene (T), or flavonoid (F). [Click here to view] |
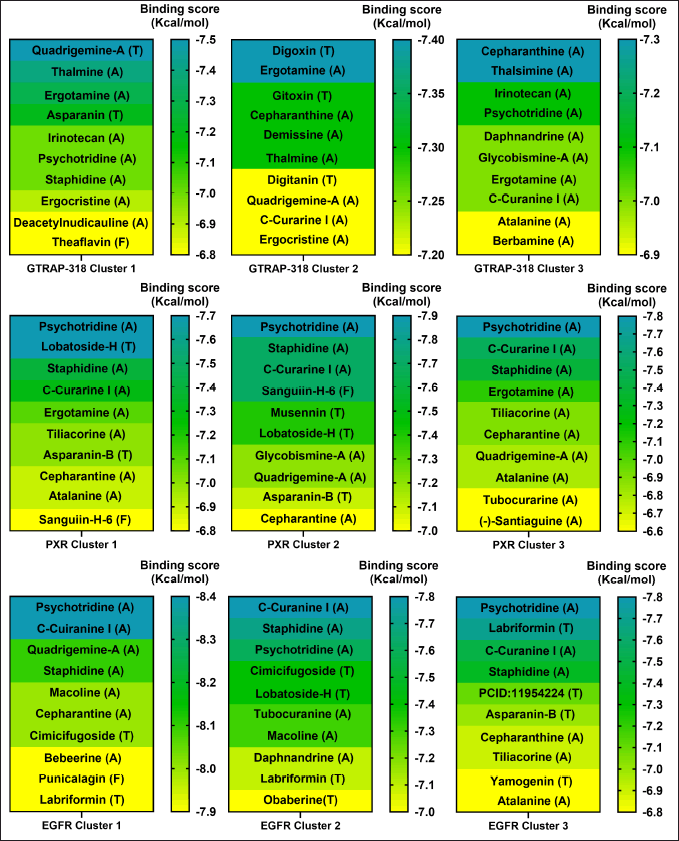 | Figure 3. Docking scores of the best 10 compounds were obtained after MDs clustering of GTRAP3-18, PXR, and EGFR. The molecular docking is based on three different clusters found on each protein. Considering ligands as alkaloid (A), terpene (T), or flavonoid (F). [Click here to view] |
As mentioned above, members of the alkaloids family are those with more interaction ability; one might conjecture that this phenomenon arises due to the presence of aromatic rings with nitrogen heteroatoms, which, in addition to being electronegative, is an electron donor to form interactions with another aromatic, and charged amino acids [44], conferring stability to the ligand inside the binding site and enhancing affinity.
On the other hand, terpenes (the second family with most of the interactions) are compounds with high lipophilicity that allows them to reach the deepest amino acids inside the protein and remain longer in the binding site, inducing conformational changes in the protein to which they have bound, making them good ligands for cytosolic proteins [45]. Polyphenols contain hydroxyl groups that can act as hydrogen bond donors and acceptors, allowing them to effectively interact with aromatic, aliphatic, acidic, and basic amino acids [46]. Despite these chemical characteristics, the results show lower binding scores than alkaloids.
Molecular docking
This approach allowed us to confirm our previous VS results, and let us identify with greater certainty the most probable ligand-protein interactions based on the analysis of 1,000 independent docking assays performed with the ligand of the highest docking score or each cluster. Representative data of the docking assay are presented in Figures 2 and 3, and Table 1.
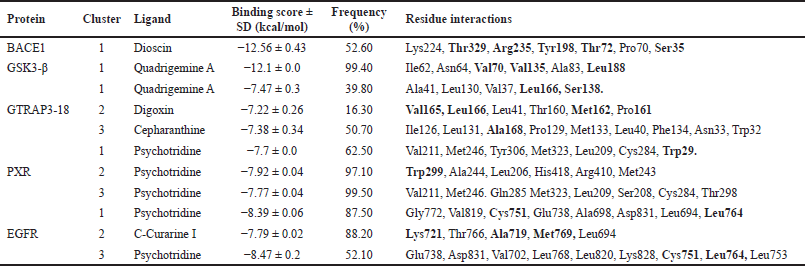 | Table 1. Results of docking assay between best phytochemicals and the clusters of the key five MCI associated proteins. Residues in bold are important for protein activity. [Click here to view] |
Regarding the active form of BACE1 at pH 4.5 (PDB ID: 2ZHT), important for APP cleavage activity in endosomes [13,47], the spirostanyl glycoside dioscin (a terpene) found mainly in Ophiopogon intermedius, and Dracaena draco showed a 12.56 ± 0.43 kcal/mol binding score (Fig. 2) on 56.2% of the independently performed assays, making a carbon-hydrogen bond with the main catalytic residues Asp228 and Asp32 [13] and hydrogen bonds with some others of the endogenous substrate binding pocket: Thr72 and Arg235 [48]. Additionally, dioscin interacts with Thr329, Arg 235, Tyr 198, Thr 72, and Ser35 (Results obtained from Molecular docking), which matches other reports [13,48,49]. This approach is supported by another report that suggests that dioscin could reduce oxidative stress and inflammation through RAGE/NOX 4 in vitro and in vivo models of Alzheimer’s disease [50]. Furthermore, our MDs analysis showed that the complex BACE1 vs dioscin remains at an inactive conformation for at least 43 ns, with slight ligand conformational differences, mainly on the glycosidic moiety of the molecule.
Additionally, focusing on GSK3β, a crucial Tau kinase enzyme, is a significant strategy for mitigating the harm caused by Alzheimer’s disease. [5], we found that the pyrrole indole alkaloid quadrigemine A, found mainly in Psychotria forsteriana, and Psychotria milnei, interact at the ATP-binding site of this kinase, with hydrogen bonds or pi-alkyl interactions with Val 70, Val 135, and Leu 188, residues that match with previous reports [51,52], where the inhibitory effect of pyrazolo[1,5-a]pyrimidin-7-amine] derivatives was tested. These interactions were found in 99.4% of the independent assays performed with a high binding score (−12.1 ± 0.1 Kcal/mol) and could be observed in Figure 2.
Our analysis and results showed the probability of three different compounds binding the C-terminus of GTRAP3-18, interacting with the first, second, and third clusters are quadrigemine A, digoxin, and cepharanthine, respectively, as shown in Figure 3. Residues involved in the mentioned interactions include Ser138, Met162, Pro161, Val165, Leu166, and Ala168, on the C-terminus, an important structure for GTRP3-18 function [17]. Except for the evidence of digoxin showing the antioxidative effect on an in vitro model of murine ischemia [53] and our theoretical approach, there is no more evidence of probable biological action in MCI or MND of these compounds.
Moreover, the best candidate for PXR binding is psychotridine, a pyrrole indole alkaloid found mainly in Psychotria forsteriana, Psychotria oleoides, and Psychotria colorata. This compound showed a mean binding score of −7.8 ± 0.05 Kcal/mol (Fig. 3), considering the three evaluated clusters, interacting with the important residues Met 246, Gln 284, Trp 299, and Tyr 306, which comprises the ligand binding domain of this nuclear receptor and match some of those reported for garcinoic acid, a selective PXR agonist [54].
Finally, respecting EGFR vs Psychotridine we obtained a −8.39 ± 0.06 Kcal/mol for cluster 1 in 87.5% of the independent assays and −8.47 ± 0.2 for cluster 3 in 51.2% of the evaluated assays, as it is shown in Figure 3. Both, show interactions with the key residues Cys751 and Leu764, which are part of the ATP-binding pocket, a common therapeutic target site for EGFR inhibitors [55] which represent suppression of reactive astrocytes, reduction of amyloid-β toxicity and neuroinflammation, an increase of axonal regeneration, and reduction of microtubule damage in neurodegenerative disorders [56]. On the other hand, the highest affinity ligand in cluster 2 was C-Curarine I with −7.79 ± 0.02 kcal/mol affinity in 82% of the 1,000 docking independent assays (Fig. 3). C-Curarine I is another alkaloid found in curare, which is an extract of many plants, including Chondrdendron tomentosum; the natives of South America used this extract as poison for hunting, affecting muscle contraction control and have very low LD50 (0.22 mg/kg) in female rats [57] whereby this molecule could be discarded as a candidate to test in MCI or MND animal models.
Protein-ligand MDs
To find the most representative conformation of the protein-ligand complex, confirm the interactions found in the docking assays are preserved during simulation time, and evaluate the stability of the complex, a cluster analysis was performed with a 0.25 Å cut-off for each complex, and the three major conformations for each one were considered for further analysis. In this sense, after 79.3 ns, BACE1 and dioscin reach the best conformation, preserved for 17.18 ns of the whole dynamics. A representative image of the interactions formed in this complex conformation is shown in Figure 4; the same analysis for the other two representative conformations of this complex corresponds to 13.89 and 11.73 ns of whole dynamics.
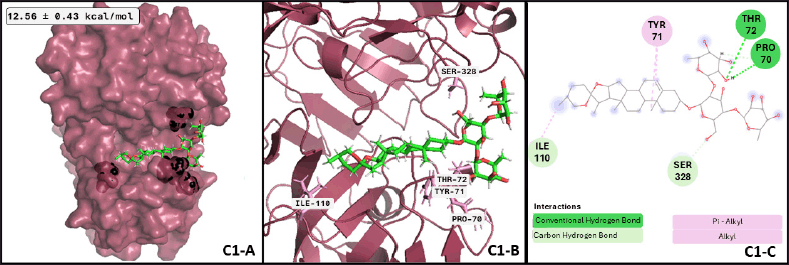 | Figure 4. BACE1-dioscin complex after MDs: (C1-A) Best conformation of BACE1 (raspberry-red), dioscin (green), and the interacting atoms in black spheres. (C1-B) 3D view of interacting atoms with labeled residues (pink). (C1-C) interacting atoms and interaction type in a 2D scheme (Conventional hydrogen bond in green, carbon-hydrogen bond in light green, and alkyl interactions in pink). [Click here to view] |
Regarding GSK3β and quadrigemine A, the best conformation of the complex is reached at 0.38 ns, with a duration of 49.67 ns, almost half of the whole dynamics. The successive two conformations have a duration of 19.54 and 8.79 ns, respectively. These three conformations represent 78% of the dynamics. Analyzing the differences between ligand conformation, cluster 1 and 3 are too similar; cluster 2 has a slight displacement to the right side of the protein, anchoring one of the tricycles inside the protein. The image of the representative conformation is shown in Figure 5, which also represents the formed interactions that were shared with the results of the previous docking assay. However, there is no previous evidence of quadrigemine A probed as a probable treatment for MCI or MND. According to our MD results, quadrigemine-A promotes the inactive form of GSK3β for a minimum of 50 ns, presenting a significant opportunity for pharmacological testing.
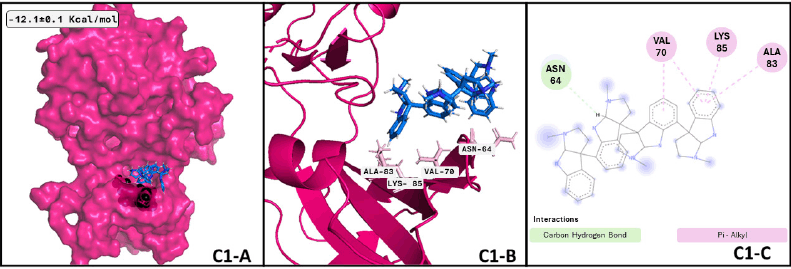 | Figure 5. GSK3β —quadrigemine A complex after MDs: (C1-A) Best conformation of GSK3β (hot pink), quadrigemine A (marine-blue), and the interacting atoms in black spheres. (C1-B) 3D view of interacting atoms with labeled residues (pink). (C1-C) Interacting atoms and interaction type in a 2D scheme (Carbon hydrogen bond in light green and pi-alkyl interactions in pink). [Click here to view] |
On the other hand, concerning GTRAP3-18 and quadrigemine-A (Fig. 6), the best conformation is achieved at 38.27 ns of the whole dynamics, with a size of 13.02 ns. The ligand has at least six transitions hanging around 3.9 to 2.9 ns; differences in the first three ligand conformations are minimal. However, these differences make quadrigemine reach other amino acids compared to molecular docking data. In the same sense, the best conformation of the complex GTRAP3-18 and digoxin in Figure 6 C2-A, C2-B, and C2-C has a size of 11.09 ns, which is reached at about 10.34 ns with at least five sudden transitions to clusters with a low size (approximately 1.5 ns); nevertheless, the best conformation forms an interaction with four of the residues that we showed on the molecular docking analysis; minor conformational differences are shown between the three first clusters. Finally, regarding the best conformation of GTRAP3-18 and cepharanthine is adopted at 36.5 ns, covering 40.06 ns of the whole dynamics; the following two conformations adopted represent 14.93 and 6.58 ns, respectively, those conformations are also close to the initial binding site proposed by the molecular docking analysis, although the amino acids that form the interactions are not the same, MDs give us a more realistic approach and the interactions are more reliable after this analysis performance. Regulation of GTRP3-18 can contribute to avoiding MCI progression because this protein is a key down regulator of glutathione synthesis, whose blocking results in a decrease of plasma glutathione levels during MCI and Alzheimer’s disease [58], promoting oxidative damage in neurons.
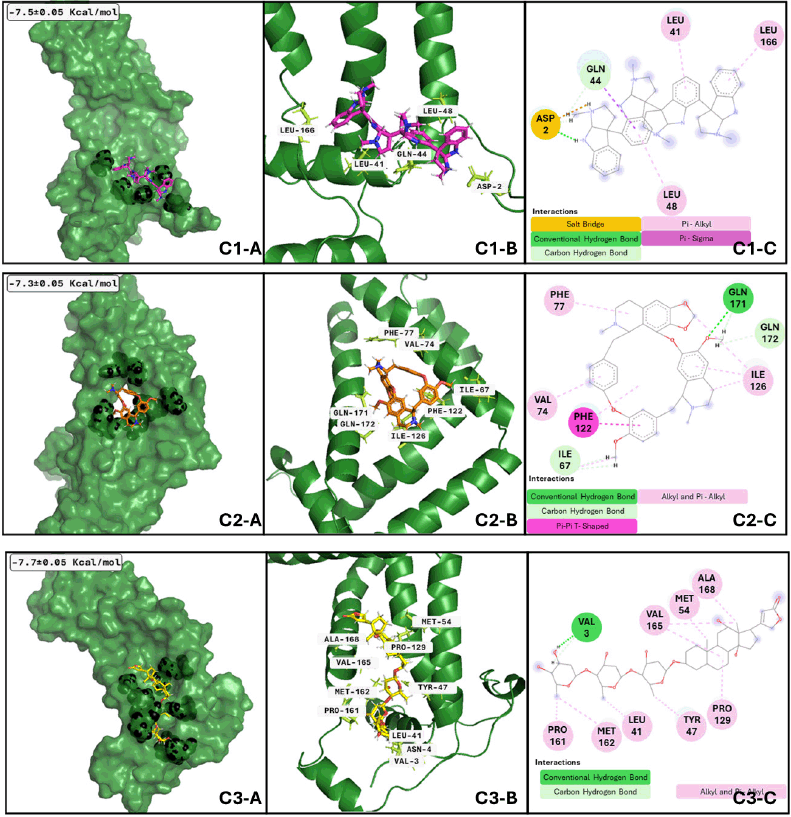 | Figure 6. GTRAP3-18 clusters after MDs: Cluster 1) (C1-A) Best conformation of GTRAP3-18 (green)- quadrigemine A (hotpink) and interacting atoms in black spheres. (C1-B) 3D view of interacting atoms with labeled residues (lemon). (C1-C) interacting atoms and interaction type in 2D. Cluster 2) (C2-A) Best conformation of GTRAP-318 (green)- digoxin (yellow) and interacting atoms in black spheres. (C2-B) 3D view of interacting atoms with labeled residues (limon). (C2-C) interacting atoms and interaction type in 2D. Cluster 3) (C3-A) Best conformation of GTRAP3-18 (green)- cepharanthine (orange) and interacting atoms in black spheres. (C3-B) 3D view of interacting atoms with labeled residues (limon). (C3-C) interacting atoms and interaction type in 2D. [Click here to view] |
Three clusters of PXR and two conformations of psychotridine (Fig. 7 C3-A, C3-B, and C3-B) dominate the interaction phenomenon; these conformations have a size between 72 and 65 ns of the whole dynamics, with at least six significant clusters along the performed dynamics. The second conformation varies between 18.5 and 16 ns. These facts reflect the stability of the complex formed between the different conformations of PXR and the ligand and the formation of important interactions inside the hydrophobic cavity of the ligand binding domain of the receptor. Among the analyzed proteins, PXR is the sole receptor that interacts with just one ligand in all three of its suggested conformations. Finding an agonist of PXR could improve memory deficit in MNDs, such as meclizine [59], and could regulate p-glycoprotein in BBB [20], which has an important role in beta-amyloid clearance. In this way, our results show psychotridine as a potential PXR agonist to be assessed in MCI models or major neurocognitive models. The active conformation of the ligand binding domain of PXR is prompted by psychotridine for an average time of 68 ns, the highest value from the evaluated compounds of this work, this information lets us propose this compound as a potent agonist for this nuclear receptor.
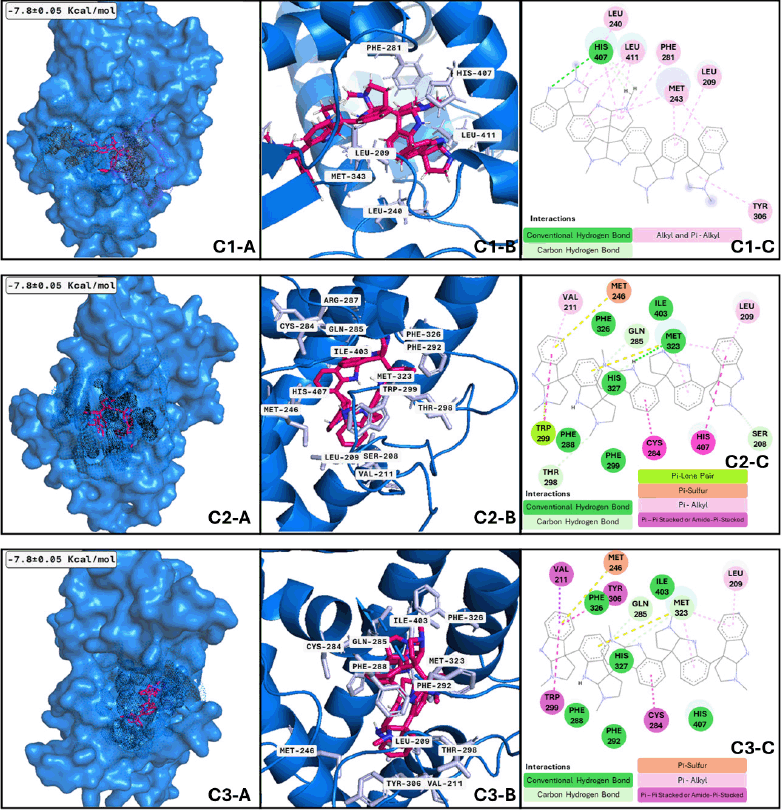 | Figure 7. PXR clusters after MDs: Cluster 1) (C1-A) Best conformation of PXR (marine-blue)-psychotridine (hotpink) and interacting atoms in black dots. (C1-B) 3D view of interacting atoms with labeled residues (light blue). (C1-C) interacting atoms and interaction type in 2D. Cluster 2) Applies the same description of PXR-psychotridine but is shown in squares C2-A, C2-B, and C2-C using the same colors but different interactions. Cluster 3) Applied the same description of PXR-psychotridine but in squares C3-A, C3-B, and C3-C in the same colors but with different interactions. [Click here to view] |
Regarding the analysis of EGFR and psychotridine (Fig. 8), the best-adopted conformation considered the most stable and representative of the binding phenomenon, is size 20.46 ns and is reached by the nanosecond nine. A few transitions to conformations with less than 1.5 ns were presented during the adoption of this conformation. The following clusters presented have a size of 19.72 and 17.99 ns, respectively. Between these three conformations, the central part of the molecule plays a significant role in the formation of interaction and anchoring to the binding site due to the molecular feature, which has fewer movements. Similarly, EGFR and C-curarine I (Fig. 8 C2-A, C2-B, and C3-B) cluster analysis showed us a great number of conformations and several transitions between them; considering only those clusters that remain for more than 1 ns, we found 26 conformations.
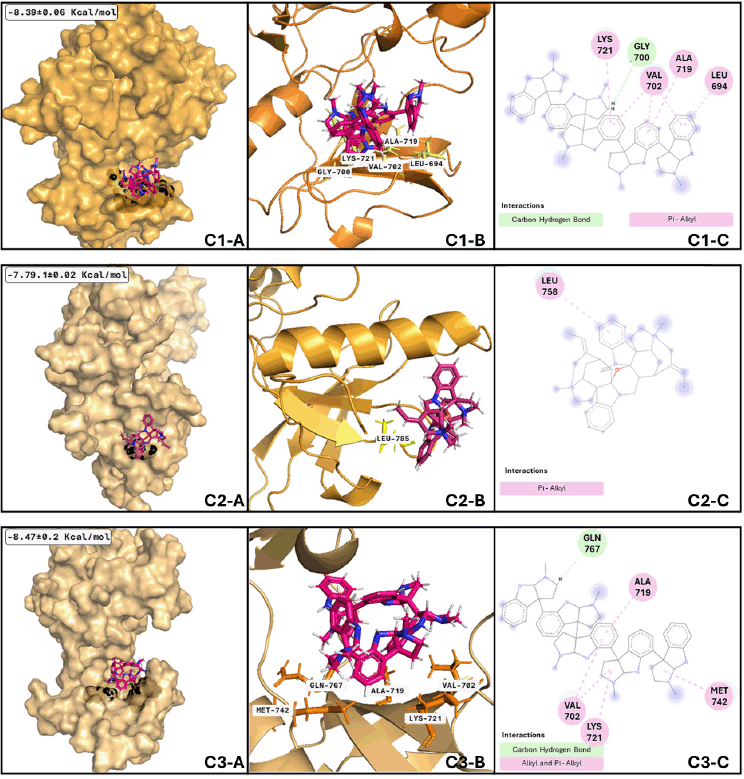 | Figure 8. EGFR clusters after MDs: Cluster 1) (C1-A) Best conformation of EGFR (bright orange)-psychotridine (hotpink) and interacting atoms in black spheres. (C1-B) 3D view of interacting atoms with labeled residues (yellow). (C1-C) interacting atoms and interaction type in 2D. Cluster 2) (C2-A) Best conformation of EGFR (light orange)- C-curarine-I (hotpink) and interacting atoms in black spheres. (C2-B) 3D view of interacting atoms with labeled residues (yellow). (C2-C) interacting atoms and interaction type in 2D. Cluster 3) (C3-A) Best conformation of EGFR (light orange)-psychotridine (hotpink) and interacting atoms in black spheres. (C3-B) 3D view of interacting atoms with labeled residues (orange). (C3-C) interacting atoms and interaction type in 2D. [Click here to view] |
Nevertheless, the most stable conformation (those with the biggest size) is reached near nanosecond thirty-three and remains during 5.8 ns of the whole dynamics; this means that most of the interacting residues are not like those shown with the molecular docking analysis. Talking about EGFR and psychotridine in the third analyzed cluster (Fig. 8 C1-A, C1-B, and C1-B), six conformations represent 50% of the whole dynamics, and the best conformation remains at 14.59 ns and reaches nearly 53 ns. The most stable and representative conformations include 10.54 and 8.22 ns, respectively. Molecular analysis of those conformations shows that the ligand stays at the initial proposed binding site, with not several differences between the main chain of psychotridine.
CONCLUSION
This study sheds light on potential therapeutic avenues for MCI treatment, emphasizing the significance of phytochemicals in modulating key molecular pathways associated with cognitive decline, letting us propose dioscin, quadrigemine-A, psychotridine, and cepharanthine, as an important candidate for be tested in MCI and Alzheimer’s models (in vitro and in vivo).
ACKNOWLEDGMENTS
Cristian Gonzalez-Ruiz would like to thank the Post-Doctoral Fellowship Program of UNAM (Programa de becas posdoctorales en la UNAM), assigned in Facultad de Estudios Superiores Iztacala by DGAPA, Tlalnepantla, Estado de México, México.
Isabel Hidalgo would like to thank the Post-Doctoral Fellowship Program of (Consejo Nacional de Humanidades Ciencia y Tecnología) CONAHCyT, México.
Miguel Ortiz-Flores would like to thank the Post-Doctoral Fellowship Program of (Consejo Nacional de Humanidades Ciencia y Tecnología) CONAHCyT, México.
AUTHORS’ CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
CONFLICT OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
The authors declare that all the data supporting the findings of this study are available within the paper.
PUBLISHER’S NOTE
All claims expressed in this article are solely those of the authors and do not necessarily represent those of the publisher, the editors and the reviewers. This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
USE OF ARTIFICIAL INTELLIGENCE (AI)-ASSISTED TECHNOLOGY
The authors declares that they have not used artificial intelligence (AI)-tools for writing and editing of the manuscript, and no images were manipulated using AI.
REFERENCES
1. Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, Kokmen E. Mild cognitive impairment. Arch Neurol. 1999 Mar 1;56(3):303.
2. Jongsiriyanyong S, Limpawattana P. Mild cognitive impairment in clinical practice: a review article. A J Alzheimer’s Dis Other Dementiasr. 2018 Dec 1;33(8):500–7.
3. Mufson EJ, Binder L, Counts SE, DeKosky ST, deToledo-Morrell L, Ginsberg SD, et al. Mild cognitive impairment: pathology and mechanisms. Acta Neuropathol. 2012 Jan 19;123(1):13–30.
4. Shen Y, Wang H, Sun Q, Yao H, Keegan AP, Mullan M, et al. Increased plasma beta-secretase 1 may predict conversion to Alzheimer’s disease dementia in individuals with mild cognitive impairment. Biol Psychiatry. 2018 Mar;83(5):447–55.
5. Forlenza O V., Torres CA, Talib LL, de Paula VJ, Joaquim HPG, Diniz BS, et al. Increased platelet GSK3B activity in patients with mild cognitive impairment and Alzheimer’s disease. J Psychiatr Res. 2011 Feb;45(2):220–4.
6. Kasper S, Bancher C, Eckert A, Förstl H, Frölich L, Hort J, et al. Management of mild cognitive impairment (MCI): the need for national and international guidelines. World J Biol Psychiatry. 2020 Sep 13;21(8):579–94.
7. Zhang J, Wang Y, Dong X, Liu J. Crocetin attenuates inflammation and amyloid-β accumulation in APPsw transgenic mice. Immunity Ageing. 2018 Dec 30;15(1):24.
8. Desideri G, Kwik-Uribe C, Grassi D, Necozione S, Ghiadoni L, Mastroiacovo D, et al. Benefits in cognitive function, blood pressure, and insulin resistance through cocoa flavanol consumption in elderly subjects with mild cognitive impairment. Hypertension. 2012 Sep;60(3):794–801.
9. Boespflug EL, Eliassen JC, Dudley JA, Shidler MD, Kalt W, Summer SS, et al. Enhanced neural activation with blueberry supplementation in mild cognitive impairment. Nutr Neurosci. 2018 Apr 21;21(4):297–305.
10. Kumar Gp, Khanum F. Neuroprotective potential of phytochemicals. Pharmacogn Rev. 2012;6(12):81.
11. Yan R, Vassar R. Targeting the β secretase BACE1 for Alzheimer’s disease therapy. Lancet Neurol. 2014 Mar;13(3):319–29.
12. Hampel H, Vassar R, De Strooper B, Hardy J, Willem M, Singh N, et al. The β-Secretase BACE1 in Alzheimer’s Disease. Biol Psychiatry. 2021 Apr;89(8):745–56.
13. Shimizu H, Tosaki A, Kaneko K, Hisano T, Sakurai T, Nukina N. Crystal Structure of an Active Form of BACE1, an Enzyme Responsible for Amyloid β Protein Production. Mol Cell Biol. 2008 Jun 1;28(11):3663–71.
14. Chen G fang, Xu T hai, Yan Y, Zhou Y ren, Jiang Y, Melcher K, et al. Amyloid beta: structure, biology and structure-based therapeutic development. Acta Pharmacol Sin. 2017 Sep 17;38(9):1205–35.
15. Zhang X, Song W. The role of APP and BACE1 trafficking in APP processing and amyloid-β generation. Alzheimers Res Ther. 2013;5(5):46.
16. Muralidar S, Ambi SV, Sekaran S, Thirumalai D, Palaniappan B. Role of tau protein in Alzheimer’s disease: the prime pathological player. Int J Biol Macromol. 2020 Nov;163:1599–617.
17. Aoyama K, Nakaki T. Inhibition of GTRAP3-18 may increase neuroprotective glutathione (GSH) synthesis. Int J Mol Sci. 2012 Sep 20;13(12):12017–35.
18. Watabe M, Aoyama K, Nakaki T. A dominant role of GTRAP3-18 in neuronal glutathione synthesis. J Neurosci. 2008 Sep 17;28(38):9404–13.
19. Chai AB, Leung GKF, Callaghan R, Gelissen IC. P-glycoprotein: a role in the export of amyloid-β in Alzheimer’s disease? FEBS J. 2020 Feb 9;287(4):612–25.
20. Ott M, Fricker G, Bauer B. Pregnane X Receptor (PXR) regulates P-glycoprotein at the blood-brain barrier: functional similarities between pig and human PXR. J Pharmacol Exp Therap. 2009 Apr;329(1):141–9.
21. Hussain B, Fang C, Chang J. Blood–brain barrier breakdown: an emerging biomarker of cognitive impairment in normal aging and dementia. Front Neurosci. 2021 Aug 19;15:688090.
22. Tavassoly O, Sato T, Tavassoly I. Inhibition of brain epidermal growth factor receptor activation: a novel target in neurodegenerative diseases and brain injuries. Mol Pharmacol. 2020 Jul;98(1):13–22.
23. Romano R, Bucci C. Role of EGFR in the nervous system. Cells. 2020 Aug 12;9(8):1887.
24. Kim S, Chen J, Cheng T, Gindulyte A, He J, He S, et al. PubChem in 2021: new data content and improved web interfaces. Nucleic Acids Res. 2021 Jan 8;49(D1):D1388–95.
25. Neveu V, Perez-Jimenez J, Vos F, Crespy V, du Chaffaut L, Mennen L, et al. Phenol-explorer: an online comprehensive database on polyphenol contents in foods. Database. 2010 Jul 30;2010(0):bap024–bap024.
26. Harun S, Abdullah-Zawawi MR, A-Rahman MRA, Muhammad NAN, Mohamed-Hussein ZA. SuCComBase: a manually curated repository of plant sulfur-containing compounds. Database. 2019 Jan 1;2019: baz021.
27. Hastings J, Owen G, Dekker A, Ennis M, Kale N, Muthukrishnan V, et al. ChEBI in 2016: improved services and an expanding collection of metabolites. Nucleic Acids Res. 2016 Jan 4;44(D1):D1214–9.
28. Blaževi? I, Montaut S, Bur?ul F, Olsen CE, Burow M, Rollin P, et al. Glucosinolate structural diversity, identification, chemical synthesis and metabolism in plants. Phytochemistry. 2020 Jan;169:112100.
29. Sterling T, Irwin JJ. ZINC 15 – Ligand discovery for everyone. J Chem Inf Model. 2015 Nov 23;55(11):2324–37.
30. O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open babel: an open chemical toolbox. J Cheminform. 2011 Dec 7;3(1):33.
31. Burley SK, Bhikadiya C, Bi C, Bittrich S, Chen L, Crichlow G V, et al. RCSB protein data bank: powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 2021 Jan 8;49(D1):D437–51.
32. Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021 Aug 26;596(7873):583–9.
33. Varadi M, Anyango S, Deshpande M, Nair S, Natassia C, Yordanova G, et al. AlphaFold protein structure database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022 Jan 7;50(D1):D439–44.
34. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004 Oct;25(13):1605–12.
35. Berendsen HJC, van der Spoel D, van Drunen R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput Phys Commun. 1995 Sep;91(1–3):43–56.
36. Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, et al. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015 Sep;1–2:19–25.
37. Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009 Dec 27;30(16):2785–91.
38. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010 Jan 30;31(2):455–61.
39. Eberhardt J, Santos-Martins D, Tillack AF, Forli S. AutoDock Vina 1.2.0: New docking methods, expanded force field, and python bindings. J Chem Inf Model. 2021 Aug 23;61(8):3891–8.
40. Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, et al. CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem. 2010 Mar 2;31(4):671–90.
41. Yu W, He X, Vanommeslaeghe K, MacKerell AD. Extension of the CHARMM general force field to sulfonyl-containing compounds and its utility in biomolecular simulations. J Comput Chem. 2012 Dec 5;33(31):2451–68.
42. Pymol Schrodinger LLC. The PyMOL Molecular Graphics System. 2015.
43. Dassault Systèmes. BIOVIA DISCOVERY STUDIO. 2021.
44. Larrañaga O, Miranda JI, Cossío FP, Cózar A de. Alkaloids reactivity: DFT analysis of selective demethylation reactions. J Org Chem. 2018 Dec 21;83(24):15101–9.
45. Cox-Georgian D, Ramadoss N, Dona C, Basu C. Therapeutic and medicinal uses of terpenes. In: Medicinal plants. Cham, Swizterland: Springer International Publishing; 2019. pp. 333–59.
46. Yilmaz H, Gultekin Subasi B, Celebioglu HU, Ozdal T, Capanoglu E. Chemistry of protein-phenolic interactions toward the microbiota and microbial infections. Front Nutr. 2022 Jul 1;9:914118.
47. Walter J, Fluhrer R, Hartung B, Willem M, Kaether C, Capell A, et al. Phosphorylation regulates intracellular trafficking of β-secretase. J Biol Chem. 2001 Jan;276(18):14634–41.
48. Zou Y, Li L, Chen W, Chen T, Ma L, Wang X, et al. Virtual screening and structure-based discovery of indole acylguanidines as potent β-secretase (BACE1) inhibitors. Molecules. 2013 May 16;18(5):5706–22.
49. Hong L, Koelsch G, Lin X, Wu S, Terzyan S, Ghosh AK, et al. Structure of the protease domain of memapsin 2 (β-Secretase) complexed with inhibitor. Science (1979). 2000 Oct 6;290(5489):150–3.
50. Guan L, Mao Z, Yang S, Wu G, Chen Y, Yin L, et al. Dioscin alleviates Alzheimer’s disease through regulating RAGE/NOX4 mediated oxidative stress and inflammation. Biomed Pharmacother. 2022 Aug;152:113248.
51. Wang Y, Wach JY, Sheehan P, Zhong C, Zhan C, Harris R, et al. Diversity-oriented synthesis as a strategy for fragment evolution against GSK3β. ACS Med Chem Lett. 2016 Sep 8;7(9):852–6.
52. Lee Y, Yoon SB, Hong H, Kim HY, Jung D, Moon BS, et al. Discovery of GSK3β inhibitors through in silico prediction-and-experiment cycling strategy, and biological evaluation. Molecules. 2022 Jun 14;27(12):3825.
53. de Souza Gonçalves B, de Moura Valadares JM, Alves SLG, Silva SC, Rangel LP, Cortes VF, et al. Evaluation of neuroprotective activity of digoxin and semisynthetic derivatives against partial chemical ischemia. J Cell Biochem. 2019 Oct 16;120(10):17108–22.
54. Bartolini D, De Franco F, Torquato P, Marinelli R, Cerra B, Ronchetti R, et al. Garcinoic acid is a natural and selective agonist of pregnane X receptor. J Med Chem. 2020 Apr 9;63(7):3701–12.
55. Stamos J, Sliwkowski MX, Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J Biol Chem. 2002 Nov;277(48):46265–72.
56. Mansour HM, Fawzy HM, El-Khatib AS, Khattab MM. Potential repositioning of anti-cancer EGFR inhibitors in Alzheimer’s disease: current perspectives and challenging prospects. Neuroscience. 2021 Aug;469:191–6.
57. Maurya SK, Periasamy M, Bal NC. High gender -specific susceptibility to curare- a neuromuscular blocking agent. Biol Res. 2013;46(1):75–8.
58. Lin CH, Lane HY. Plasma glutathione levels decreased with cognitive decline among people with mild cognitive impairment (MCI): a two-year prospective study. Antioxidants. 2021 Nov 19;10(11):1839.
59. Singh H, Sodhi RK, Chahal SK, Madan J. Meclizine ameliorates memory deficits in streptozotocin-induced experimental dementia in mice: role of nuclear pregnane X receptors. Can J Physiol Pharmacol. 2020 Jun;98(6):383–90.