
INTRODUCTION
The complexity of cancer has provided both researchers and healthcare professionals with a significant opportunity to advance understanding and treatment. Across various classifications, cancer remains a leading cause of death worldwide [1], despite the implementation of multiple therapeutic approaches. One of the most daunting challenges lies in the intricate etiology of cancer, particularly as it progresses to advanced stages. The widely accepted tumor node metastasis staging system underscores the complexity of cancer diagnosis and treatment planning. While early-stage tumors may be effectively managed through surgical interventions, the presence of metastasis necessitates a combination of adjuvant chemo radiation and surgical modalities. However, the success rates of these interventions remain dishearteningly low, largely due to the aggressive nature of metastatic cancer, which can infiltrate and affect multiple organs in various ways. Chemo radiation therapy, in NSCLC, paclitaxel in chemotherapy with radiation, and oxaliplatin in chemotherapy with radiation although integral to cancer treatment, often fall short of fully addressing metastatic cancer. Consequently, chemotherapy emerges as a cornerstone modality in the management of metastasized cancer. However, the emergence of chemotherapeutic drug resistance poses a significant hurdle to achieving successful treatment outcomes [2].
In recent years, the development of novel drug candidates utilizing sophisticated drug design approaches and considering explicit clinical parameters has shown promise. Despite these advancements, the persistence of chemotherapy resistance highlights the ongoing challenge of achieving a comprehensive cure for cancer. In navigating this landscape, it is crucial to delve deeper into the mechanisms driving cancer progression and treatment resistance. By embracing a multifaceted approach that combines innovative therapeutic modalities with a nuanced understanding of cancer biology, we can strive towards more effective treatment strategies and improved outcomes for patients battling this formidable disease.
Approaches on the robust basis of physiology via killing cancerous cells by oxidized free radicals and reactive oxygen species (ROS) in cells. The study revealed platinum class alkylating chemotherapeutic drug generated free radicals in cells that accept and transfer electrons oxygen and kill cancerous cells [1]. Cell organelle has a vital role involving mitochondria, endoplasmic reticulum, lysosomes, and Golgi bodies. Mitochondria and HIF have a strong interconnection in regulating cell death. Subdue mitochondrial function by HIF to participate in drug treatment failure [2].
The Otto Warburg illustrates damaged respiration and excessive fermentation in cells responsible for Cancer [3,4]. The D.N.A. of cell damage by ultra violet radiation. Carcinogens consequently defect in apoptosis liable for tumorigenesis [5]. Apoptosis of damaged D.N.A. control by (Ataxia telangiectasis mutated and RAD3 related) A.T.R./Chk-1 but mechanism not yet well established [6,7]. Evidently, upon D.N.A. damage by U.V. simultaneously activation of (Ataxia telangiectasis mutated) A.T.M./Chk-1 signaling pathway consequently down-regulates Smurf-1 and increases Rho-B consequently, apoptosis [8–10]. Failures are considered as an opportunity to be emphasized in this article. Multiple transduction pathways are to be found, analyzed, and then designed for chemotherapy. Among all other protein kinases, the most acceptable pathway is still a significant impediment in clinical oncology. Protein kinase involves receptor and non-receptor tyrosine kinase, Serine and threonine kinase, and other associated pathways. All signal transduction pathways majorly inhibit translation by altered transcription and replication whereas this perspective emphasizes hypoxia and immunotherapy. Another, parameter, tumor microenvironment (TME), was altered drug penetration due to adverse pharmacokinetic conditions consequently, promote tumor growth. Stromal cells in T.M.E. participate in uncontrolled proliferation, angiogenesis, and metastasis with drug impediments [11–18]. Tumor cells participate in hypoxia in the TME and its major impediment to cancer drugs [12]. Hypoxia participates by HIF-α and acidic TME. In hypoxic conditions, the fast proliferation of cancerous cells due to creates new angiogenic vasculature that contributes to delivering oxygenated blood [19,20]. The slowly proliferating cancer cells, under hypoxic conditions, escape from chemotherapy drugs, and these cancer cells ensure epithelial to mesenchymal transition. Some studies revealed in the acidic environment of cells lysosomes and endosomes in tumor cells support metastasis by activating protease [21,22]. The degradation of tumor cells in a neutral environment and simultaneously, improved by cytotoxic T lymphocyte, immunotherapy [23]. In the light of above point, hypoxia alters apoptosis cell death, autophagy to remove dead from cells, and immunotherapy to manage cancer.
Hypoxia bolstering ROS to induced cancer
During hypoxic conditions, ROS levels increased due to decreased oxygen utilization, the passage of electrons from the mitochondrial complex by the electron transport chain (ETC), and electron leak from the ETC, consequently overproduction of ROS. ROS destabilize genomic stability and disrupt the DNA repair pathways and simultaneously, ROS. cause mutation and promotion of cancer with multidrug resistance cells [24–27]. Hypoxic stress aids tumor resistance with immunosuppression by tumor-associated macrophages [28,29]. The drug does not diffuse into the cell in an acidic environment and simultaneously, genetic alteration in P53 due to Hypoxia, also known as ion trapping [30]. In hypoxic conditions, multidrug resistance has increased the activity of multidrug transporter p-glycoprotein (p-gp) and its expression with the mechanism of multidrug resistance expressed by the MDR1 gene. Simultaneously, in hypoxic conditions mRNA levels remain the same, and activity increases consequently, tumor progression increases [31–34]. Tumor cells in the human body then alter the metabolism of normal cells under hypoxic conditions and proliferate by capturing the host immune system, disrupting apoptosis [35,36] by Caspase-mediated selective cleavage off a subset of cellular polypeptides. Simultaneously, participating biochemical and morphological cell apoptosis [37]. DNA damage and microtubule disruption by intracellular caspase cascades accelerate by death receptor ligand system and cellular stress. DNA damage and microtubule disruption regulate B cell lymphoma-2(Bcl-2) family members have pro-apoptic and anti-apoptic class. Intracellular caspase cascade is regulated by the Bcl-2 family by cytochrome-c and other polypeptides [38,39].
Hypoxia modulation to manage Cancer
Hypoxia is to be considered as targeted for cancer therapy. Multiple ways to target hypoxic tumors are hypoxia-activated prodrug [40], hypoxia-inducible factor 1-alpha (HIF-1α) modulator, Prolyl 4-hydroxylase alpha-1 (P4HA1), Prolyl hydroxylase domain 2, Gene therapy, specific target pathways critical in hypoxia such as mTOR (mammalian target of rapamycin), unfolding protein response (UPR) pathways, Acid induced tumor. HIF-1α induced chemotherapeutic resistance in pancreatic cancer via up-regulation of cytidine triphosphate synthase and transketolase while using digoxigenin to halt HIF-1α induced translation. Consequently, encouraging results of gemcitabine in pancreatic Cancer [41–43].P4HA1 up regulated by HIF-1α in the TME of breast cancer. P4HA1 regulates cell metabolism and enhances tumors; thus, P4HA1 is a prominent target for breast cancer [41]. HIF-1α regulates the transcriptional regulation by gene TFP1 in breast cancer. Another prominent target of HIF-1α is prolyl hydroxylase domain 2 (PHD2) for breast cancer treatment. The hypoxia is further illustrated in Figure 1.
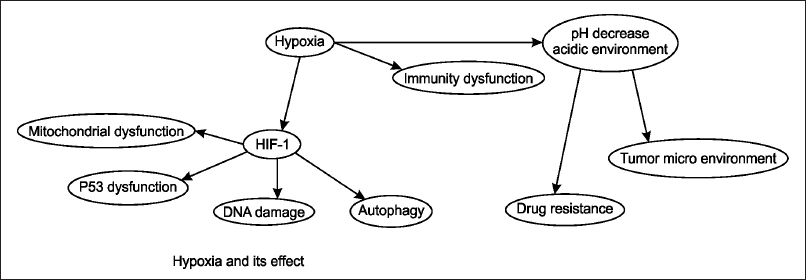 | Figure 1. The figure depicts the effects of hypoxia on cellular and physiological processes. Hypoxia leads to the activation of HIF-1αsss, which results in mitochondrial dysfunction, p53 dysfunction, DNA damage, and autophagy. Additionally, hypoxia causes a decrease in pH, creating an acidic environment that contributes to immunity dysfunction, alteration of the TME, and increased drug resistance. This interplay highlights the critical role of hypoxia and HIF-1 in tumor progression and resistance to therapy. [Click here to view] |
The hypoxic prodrug was activated by cellular reductase, re-oxidized into initial drug progenitors in anorexic cells, and then converted into a cytotoxic substance. Clinical trial phase-II results encourage hypoxic progenitor TH-302 with gemcitabine. Hypoxic agents as progenitors, also known as alkylating agents, are in Figure 2. TH-302 ((1) Evofosfamide) chemical class of alkylating agent and, in combination with (2) gemcitabine, synergistic action in pancreatic cancer [44]. Synergistic action of (3) praziquantel use as an adjunct therapy in bladder cancer with (4) Mitomycin-derived prodrug [45].
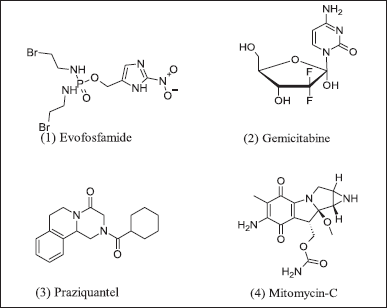 | Figure 2. Hypoxic prodrugs. [Click here to view] |
Autophagy and Cancer (Hypoxia by HIF-1α induction of autophagy)
Autophagy, a vital cellular process, is essential for maintaining cellular viability by removing misfolded proteins and dysfunctional organelles [46–52]. It is particularly crucial for cellular stress management and homeostasis, thereby inhibiting tumor formation. However, in the context of solid tumors, autophagy’s role becomes complex and controversial as it aids tumor survival under hypoxic conditions [53]. Hypoxia, often found in solid tumors, induces autophagy through the HIF-1α pathway. This pathway activates autophagy-related proteins such as ULK-1, Atg13, and FIP200 through mTOR and LKB1-AMPK signaling [54].
Autophagy is classified into three types: macroautophagy, microautophagy, and chaperone-mediated autophagy. In solid tumors, inhibiting autophagy has been shown to enhance chemotherapy effectiveness, highlighting its dual role in cancer therapy [55]. Hypoxia-induced autophagy occurs in tumor regions, where the UPR contributes to chemotherapy resistance. This resistance is mediated by the PERK-dependent transcriptional induction of microtubule-associated protein 1 light chain 3 and autophagy-related genes [55,55]. Furthermore, autophagy is associated with multidrug resistance due to the activation of ATP-binding cassette transporters. Therefore, understanding the balance between autophagy’s protective roles and its support for tumor survival under hypoxia is crucial for developing effective cancer therapies.
m-TOR pathway
The elevated level of m-TORC1 was detected in rodents and human tumors. Further, confirmed in tumor-derived cell lines have knockdown of TSC1:TSC2 [56–67]. Cell growth is regulated by the mTOR pathway involved, specifically by m-TORC1 upregulating and downregulating the TSC1/TSC2 complex. Other factors regulate cell growth by activating the m-TOR pathway and rising level A.M.P. consequently activated AMP kinase (AMPK) in multiple ways among one genotoxic stress by P-53 mediated activation. The mTORC1 activity is upregulated by growth factors through insulin or insulin-like growth factors, phosphoinositide-3’ kinase, A.K.T. pathway, Wnt-GSK3 signaling pathway and ERK-RSK kinase cascade [68,69].
m TORC1 activity modulates in adverse conditions, nutrient limitation, Hypoxia, and DNA damage disrupt the cell cycle and P53 signal. m TORC1 activity upregulates and downregulates by TSC1:TSC2 complex via GAP (GTPase activating protein) by action of GTPase-Rheb that stimulates mTORC1. m TOR regulation involves specifically sestrin-2 due to a decrease in GTP level. TSC activity also regulated by other kinase AKT, ERK, RSK, AMPK [70,71].
In stressed conditions, P53 precisely targets gene p21waf to activate sestrin, which means sestrin-1(PA26) and sestrin-2(Hi95) dimer interact with TSC1:TSC2 (Hamartin:tuberin) complex and phosphorylation by AMP induced AMPK-α consequently autophosphorylation TSC1:TSC2 complex simultaneously GAP activity by Rap1 induced by TSC2 and another factor Guanidine exchange factor also participate support by transitional controlled tumor protein study in drosophila [72] and m-TOR inhibition [73]. In cancer conditions, G.A.P. mutates, mTORC1 activation, and cell growth. m TOR proteins are serine-threonine kinase that belongs to the family of the phosphoinositide 3 kinase-related kinase and occurs in eukaryotes. m TOR occurs in two complex forms: mTORC1 and mTORC2. mTORC1 evidently participates in cancer, neurodegeneration while mTORC2 function is still not fully understood [74–77]. The m-TOR activity is illustrated in Figure 3 [78].
m TORC1 modulation was TSC complex-dependent signaling. TSC suppresses tumor cells and downregulates by phosphorylation. m-TORC1 upregulates cell growth, but the mTOR pathway still needs further research. m-TOR inhibitors (5), (6), (7), and (8) are in Figure 4. TSC complex downregulates by phosphorylation among GSK-3β, energy stress, and hypoxia to AMP activates AMPK, and hypoxia to HIF1α activates REDD1. In contrast, on the other side, hypoxia-induced HIF1α inhibited by Von Hippel-Lindau tumor suppressor furthermore WNT pathway inhibits GSK-3β to phosphorylation [79–88].
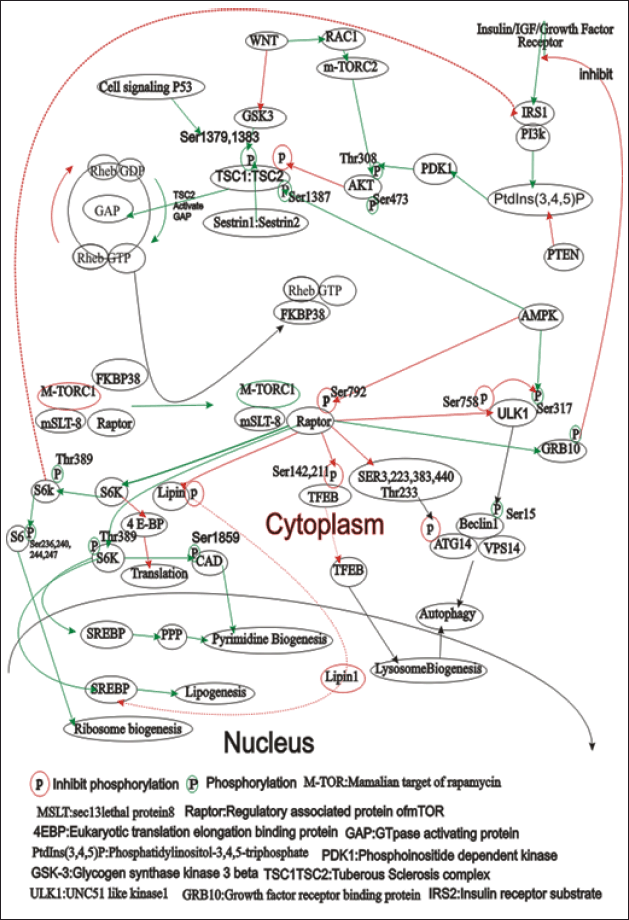 | Figure 3. The figure illustrates the complex signaling pathways involving the mammalian target of rapamycin (mTOR), focusing on its two complexes: mTORC1 and mTORC2. mTORC1 is regulated by nutrients, growth factors, and energy status, promoting protein synthesis, lipogenesis, and ribosome biogenesis while inhibiting autophagy. Key regulators include Rheb-GTP, FKBP38, TSC1/TSC2, PI3K, AKT, and AMPK. mTORC2 mainly regulates cytoskeletal organization and cell survival through AKT, SGK, and PKC. The figure also highlights the downstream effects of mTOR activation, such as translation promotion via S6K and 4EBP1, and autophagy regulation through ULK1 and Beclin1. This interplay underscores the importance of mTOR in cellular growth, metabolism, and survival, pointing to potential therapeutic targets for diseases like cancer. [Click here to view] |
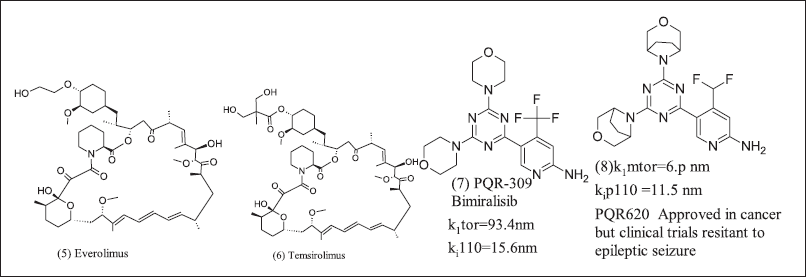 | Figure 4. mTOR modulator. [Click here to view] |
Immunotherapy and Cancer
In cancer few decades, immunotherapy is illustrated in Figure 5 as one option to combat cancer [89–91]. Immunotherapy boosts immunity to kill cancer cells at checkpoints via checkpoint inhibitors, adoptive cell transfer, and vaccines. The increased immunity combat cancer cells via several checkpoint inhibitors among Cytotoxic T- lymphocyte-associated protein-4 (CTLA-4) and programmed cell death protein 1 (PD-1) and other renowned checkpoint inhibitor monoclonal antibodies (mAB) with the disadvantage of administration schedule, time-consuming procedure and costly and toxic effects not to be altered by any way in comparison to small drug molecule. The small drug molecule can reach the target site in the TME and low immunogenicity [92]. Find out small molecules that work in the TMEt and modulate immune suppression via either activating innate immune response or adaptive immune signaling pathway to treat Cancer. Among adenosine receptor signaling at immune checkpoint prominent target. Adenosine A2AR and A2BR receptor antagonists were used for cancer management by immune-modulation. The human immune system combats pathogens by way of inflammatory response. Tumors hijack and take control of the immune system, grow, and then metastasize [93]. The immune checkpoint pathways PD1, CTLA-4, A2AR ,A2BR, TIM-3(T cell immunoglobin and mucin domain-3) [94,95]. In normal physiological conditions, the level of adenosine is less than 1 µM [96] but observed adenosine levels (10 µM) in the TME and related pathological conditions are, as Cancer related fibroblasts, regulatory T cells, myeloid-derived suppressor cells (MDSCs), endothelial cells and T helper cells [97–103].
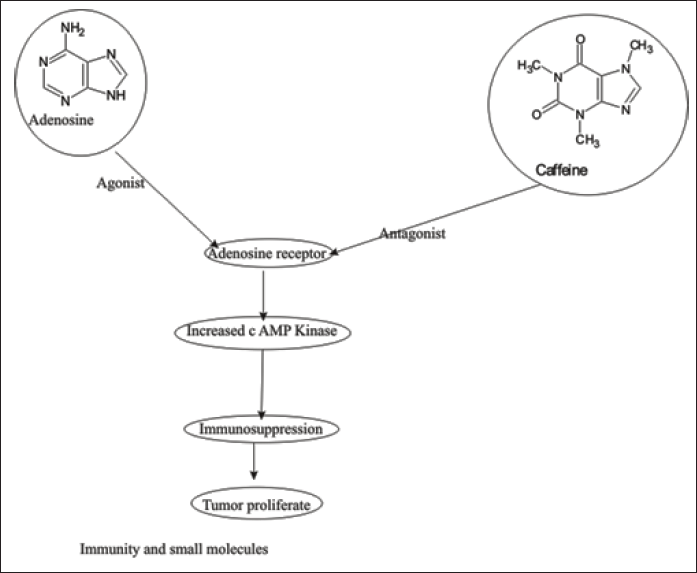 | Figure 5. The diagram illustrates the interaction between adenosine and caffeine with the adenosine receptor. Adenosine acts as an agonist, binding to the adenosine receptor, leading to an increase in cAMP kinase activity. This cascade results in immunosuppression and the promotion of tumor proliferation. Conversely, caffeine functions as an antagonist to the adenosine receptor, potentially disrupting this pathway. The overall theme is the impact of small molecules on immunity and cellular processes. [Click here to view] |
Upon binding adenosine to A2AR and A2BR to increase c AMP protein kinase, the signal is associated with the immune system and consequently, immunosuppressant in the TME. Increased intracellular c AMP level in T cells due to activation of A2AR and A2BR receptor consequently, decreased Tumor necrosis factor (TNFα), interleukin 2, and interferon Gamma, and compromised CD-8+ T cell infiltration [104–106]. Adenosine 2A receptor activation on regulatory T cells increases PD-1 and CTLA-4 expression, and macrophages promote tumor cell proliferation and increase immunosuppressant [107,108]. A2BR alters cell differentiation by the mononuclear-phagocyte system, consequently, cancer angiogenesis, tumor spread, and suppression of lymphocyte-mediated anti-tumor immunity [109]. Thus, Adenosine receptor antagonist, PD1, CTLA-4, a prominent oncotarget. Evidently, in some studies alone, A2AR antagonists were not encouraged as a result in CL8-1 melanoma cell line and RMA T cell lymphoma cell line exhibits dependent on CD8+ T cell activity but prominent observation metastasis decrease. The union of A2AR and A2BR antagonists with inhibition of PD-1 and CTLA-4 results in a prominent anticancer effect [110,111]. The A2BR inhibition has prominent anti-tumor effects in prostate cancer, slow growth of the bladder, and breast cancer with decreased cell proliferation. Furthermore, it decreases tumor growth and immune suppression due to MDSCs and inhibits vascular endothelial growth factor and angiogenesis [112–115].
Adenosine antagonist for cancer immunotherapy
Adenosine 2A receptors have significance in cancer immunotherapy such as pyrazole and pyrimidine core. That article emphasizes anti-tumor effects concerning immunity. Natural and synthetic analogs as Adenosine receptor antagonists are Xanthine and analogue, Pyrimidine analogue, Azolopyrimidine analogues, Triazolo pyrimidine analogues, 2-oxothiazole analogue, Pyrazole and Benzothiazole analogue, Benzimidazole, Benzothiazole, Quinoxalines, Amino Pyrimidines, Triazine isomers, Pyrazole linked heterocyclics and miscellaneous as illustrated in Table 1.
 | Table 1. Synthetic analogues for cancer. [Click here to view] |
Xanthine and its analogue
Xanthine and semi-synthetic analogues are illustrated in Figure 6. (9) Caffeine is a non-specific naturally occurring adenosine receptor antagonist (10) Istradefylline, a semi-synthetic xanthine analogue, have adenosine antagonist action and was initially designed to treat Parkinsonism, but (10) has revealed good result in Cancer [116] thus, as an option to repurpose for Cancer. Xanthine analogues have poor water solubility. Pyrazolo triazole Pyrimidine analogue non-xanthine adenosine 2A receptor antagonists are SCH58261, FSTP, ZM241385, and Tozadenant [117,118] (11). SCH58261 revealed the slowdown of metastasis in breast cancer by CD73 endogenously, and melanomas activate Nk cells (12). FSTP irreversible type antagonist and low efficacy due to impaired CD+ T cell differentiation and accumulation [119], (13) ZM241385 has poor solubility but inhibits lung cancer tumor metastasis [110], (14) Tozadenant enters clinical trial phase-3, but adverse events and scientists were further studies but encouraging results in breast cancer in combination with PD-1 inhibitors and mAB [105].
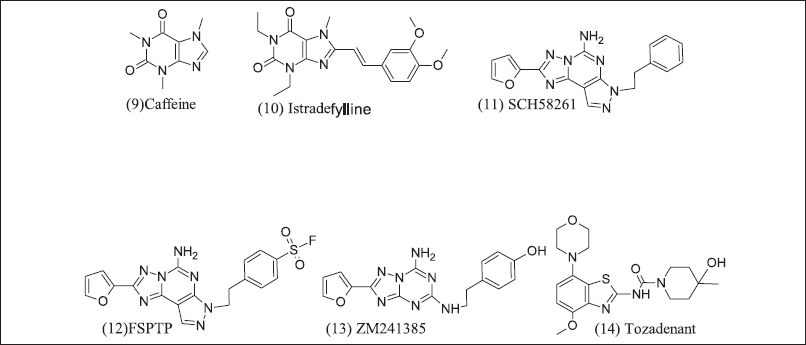 | Figure 6. Xanthine and Pyrazolo triazole pyrimidine inhibitor. [Click here to view] |
Pyrimidine analoguesAdoRx therapeutics design and patented pyrimidine derivatives are illustrated in Figure 7 as potent A2AR and A2BR antagonists. The 2-amino pyrimidine core chemical structure of the antagonist is A at R1 position furan ring and at X-R2 position aryl methylamine. These analogue (15), (16), (17), and (18) exhibit responses in lipopolysaccharide mediate TNFα release NECA reversal assay.
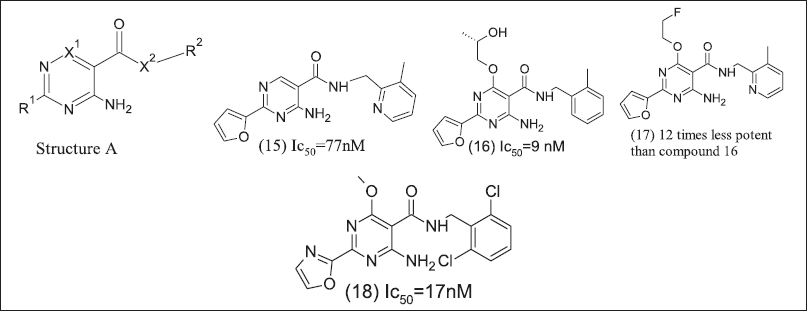 | Figure 7. Pyrimidine inhibitors. [Click here to view] |
Azolopyrimidine analogues
Arcus bioscience design and patented azolopyrimidine illustrated in Figure 8 to target Cancer [120] as an adenosine antagonist. In structure B at R4 position, substitution with methyl group activity decreases compared to hydrogen. Compound (19) exhibits A2A receptor antagonist activity in CHO-TREx cAMP assay. Compound (20) have methyl benzonitrile and dose 100 mg/kg, po, b.i.d alone [121] or in combination with anti-PD-1 (5 mg/kg) or doxorubicin(6 mg/kg), consequently anti-tumor effects on B16-F-10 melanoma [122]. Arcus further two patents, one on quinazoline pyridine as compound C and its analogues (21), (22). Another quinazoline pyrazole core compound D and its analogue (23), (24). These compounds exhibit antagonist activity as A2AR and A2BR
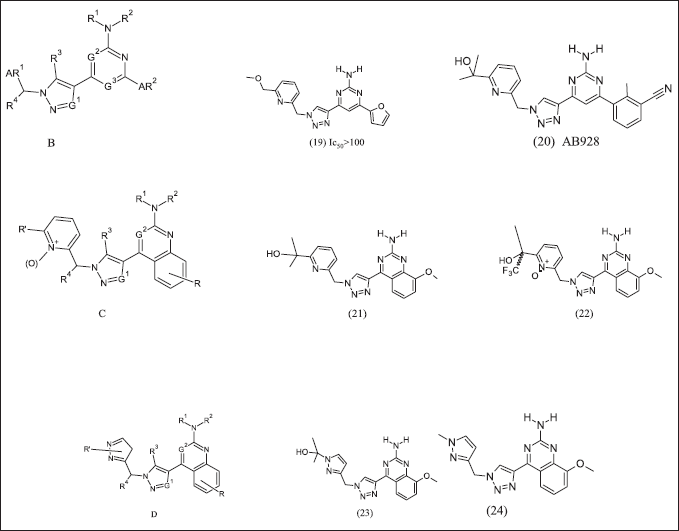 | Figure 8. Azolo pyrimidine analogues. [Click here to view] |
Triazolo pyrimidine analogues
Bristol-Myers Squibb research and design illustrated in Figure 9 (25) Vipadenant and (26) Ciforadenant have [1,2,3] triazolo[5,4-d] pyrimidine core was first introduced for Parkinsonism. It revealed the selectivity towards A2AR, but it was discontinued due to failure in the phase-2 clinical trial in preclinical cancer immunology research and observed an othosteric binding site. Ciforadenant potently inhibits c AMP in HEK-293 cells at Ic50=17nM and primary human T cells at IC50=70 nM. Ciforadenant suppresses p CREB and restores PERK levels in human cells.
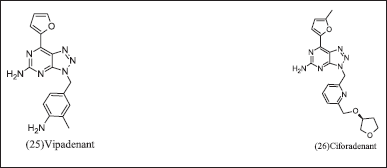 | Figure 9. Triazolo pyrimidine analogues. [Click here to view] |
2-Oxothiazole analoguei
TEAOS research group developed 2-Oxothiazole, illustrated in Figure 10, as an A2AR antagonist to target Parkinsonism’s disease, but due to a high adenosine environment, it does not exhibit antagonistic activity. The high dose required for inhibition within the tumor for cancer management [95]. E core was introduced as cAMP inhibition. It also inhibited CREB phosphorylation as a consequence of A2AR antagonism. 2-Oxothiazole and its analogues (27A, 27B).
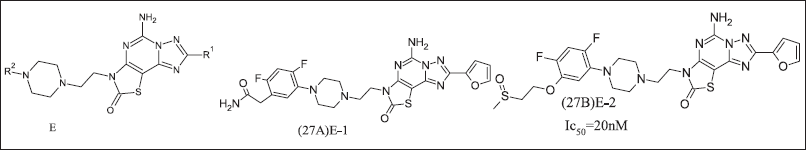 | Figure 10. Oxothiazole analogue. [Click here to view] |
Pyrazole and Benzothiazole analogue
Further researchers from Merck introduce pyrazolo [4,3-e]-1,2,4-triazolo-[1,5-c] pyrimidine analogue illustrated in Figure 11. (28) Preladenant was introduced for Parkinsonism but terminated in phase-3 consequences of failure in efficacy as compared to placebo. The Preladenant result of the phase-1 clinical trial of (NCT0399161) in advanced solid tumor but data did not support at the endpoint thus terminated. Merck has patented three Benzothiazole compounds (14) Tozadenant where nitrogen atom introduced to improve pharmacokinetic properties.
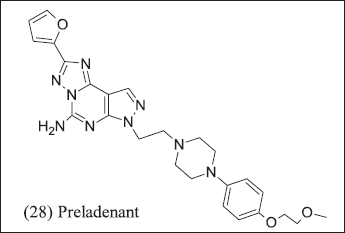 | Figure 11. Pyrazole analogue. [Click here to view] |
Benzimidazole (Bioisostere replacement of sulphur with nitrogen)
Benzimidazole and its analogue illustrated in Figure 12 have (29), (30), (31), (32), (33), and (34) A2AR antagonist effects. Benzimidazole analogues (35), (36), and (37) have the isosteric replacement of sulphur with nitrogen as a consequence of a half-life increase. The half-life of benzimidazole analogues further increased with aryl amide replacement. Consequently, the Benzimidazole morpholino group was replaced with heteroaryl, improving pharmacokinetic properties and these compounds as A2AR inhibitors. These compounds control the secretion of cytokine that is suppressed by adenosine. These compounds have IC50 = 100–1,000 nM range.
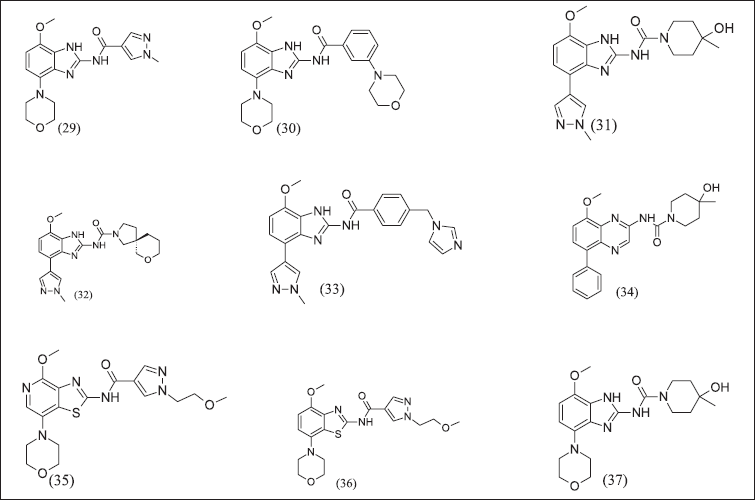 | Figure 12. Benzimidazole and isosteric replacement analogues. [Click here to view] |
Quinoxalines analogue
Quinoxalines from Benzothiazole illustrated in Figure 13 (38), (39), and (40) retained adenosine antagonistic activity and a half-life of more than 2 hours. In Quinoxalines, the analogue aryl group was replaced with tetrahydropyran, consequently decreasing half-life, and the urea side chain was replaced with aryl amide, consequently poor pharmacokinetic properties.
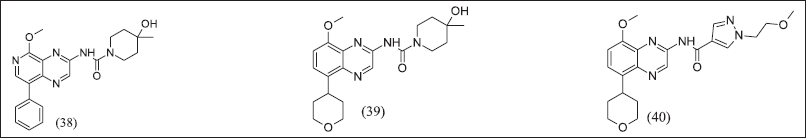 | Figure 13. Quinazoline analogue. [Click here to view] |
Amino pyrimidines
2,6-Bis(heteroaryl)-4-amino-pyrimidines design illustrated in Figure 14 and introduces Novartis/Palobiopharma as an A2AR antagonist. The potency of the compound increased by introducing an electron-withdrawing group at position 5 of the pyrimidine ring. (41) Taminadentant (PBF-509) exhibit 25 times more potent antagonism as compared to bis(hetroaryl)-4-amino pyrimidine A2AR antagonist. (42), (43) Chloro / cyano substitution at fifth position and pyrrolidyl/ethylthiol/trifluoroethoxyl at the sixth position of the bis(heteroaryl)-4-Amino pyrimidine for its antagonist response and it is important for binding to receptor. Furthermore, PBF-509 exhibits a decreased tumor burden in mice with B16-CD73+ tumors. PBF-509 with PD-L1 maintains immune response in tumor-infiltrating lymphocytes in non-small cell lung cancer cases. PBF-509 produced a synergistic anti-tumor effect with immunotherapeutic agents.
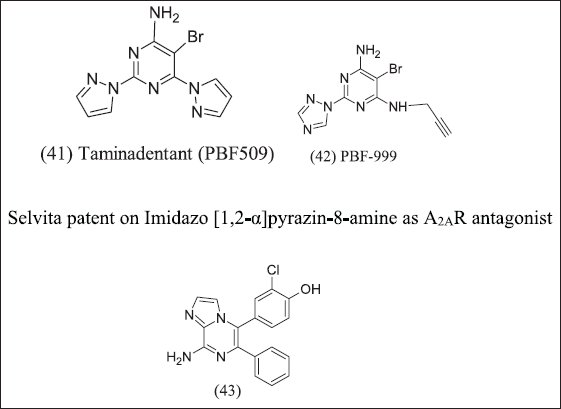 | Figure 14. Amino pyrimidine analogue. [Click here to view] |
Triazine isomers
AstraZeneca, by using computational techniques to design, illustrates in Figure 15 1,3,5-triazines isomers and 1,2,4 triazines isomers as A2AR antagonists that access ribose pocket (44), (45), (46). AZD4635 was designed by using an SBDD strategy with PDB:6GT3 and decreased tumor growth (50 mg/kg,po,b.i.d) with anti-PDL1 [123].
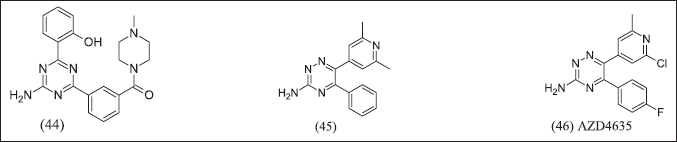 | Figure 15. Triazine analogue. [Click here to view] |
Pyrazole linked heterocyclics as a phosphodiesterase inhibitor
Overexpression of PDE-10A was observed in Cancer then, specifically, PDE-10A inhibitors illustrated in Figure 16 (47), (48), (49).
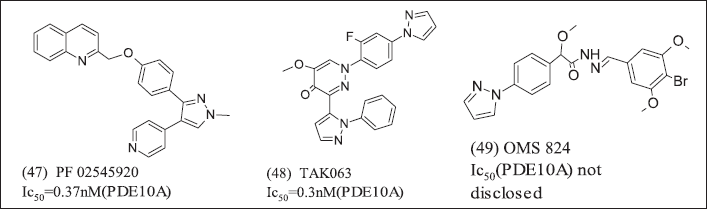 | Figure 16. Pyrazole heterocycles. [Click here to view] |
CONCLUSION
In cancer life-threatening conditions chemotherapy failure and drug resistance are major emerging areas for researchers. Hypoxia and associated conditions, tumor micro environment, ROS, and immunosuppressant condition. Adenosine receptor and hypoxia significance emphasize above and management of cancer by modulation of pathway mTOR and in immunity by CTLA 4, PD1 Benzothiazole, quinazoline, and pyrimidine analogue have prominent inhibitory responses in adenosine sites. Alkylating agents exhibit prominent effects in hypoxic tumors and drug response. Thus, design Nitrogen-containing heterocyclic, Benzothiazole, quinazoline pyrimidine analogue have both alkylating and adenosine inhibitory action.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
FINANCIAL SUPPORT
There is no funding to report.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included in this research article.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
1. Wozniak AJ, Glisson BS, Ross WE, Hande KR. Inhibition of Etoposide-induced DNA damage and cytotoxicity in L1210 cells by dehydrogenase inhibitors and other agents Cancer Res. 1984;44(2):626–32.
2. Sasabe E, Tatemoto Y, Li D, Yamamoto T, Osaki T. Mechanism of HIF-1α-dependent suppression of hypoxia-induced apoptosis in squamous cell carcinoma cells. Cancer Sci. 2005;96(7):394–402. CrossRef
3. Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–14. CrossRef
4. Kaczanowski S. Apoptosis: its origin, history, maintenance and the medical implications for cancer and aging. Phys Biol. 2016;13(3), 031001.
5. Cotter TG. Apoptosis and cancer: the genesis of a research field. Nat Rev Cancer. 2009;9:501–7. CrossRef
6. Patil M, Pabla N, Dong Z. Checkpoint kinase 1 in DNA damage response and cell cycle regulation. Cellul Mol Life Sci. 2013;70:4009–21. CrossRef
7. Hong Son B, Van Nga V, Thi Diem Hong L, Thi Quynh D. Potent natural inhibitors of Alpha-glucosidase and the application of Aspergillus spp. in diabetes type 2 drugs: a review. VNU J Sci Med Pharm Sci. 2022;38(1):002. CrossRef
8. Olcina M, Lecane PS, Hammond EM. Targeting hypoxic cells through the DNA damage response. Clin Cancer Res. 2010;16(23):5624–9. CrossRef
9. Kelly KR, Friedberg JW, Park SI, McDonagh K, Hayslip J, Persky D, et al. Phase I study of the investigational aurora A kinase inhibitor alisertib plus rituximab or rituximab/vincristine in relapsed/refractory aggressive B-cell lymphoma. Clin Cancer Res. 2018;24(24):6150–9. CrossRef
10. Spaapen R, Van Den Oudenalder K, Ivanov R, Bloem A, Lokhorst H, Mutis T. Rebuilding human leukocyte antigen class II-restricted minor histocompatibility antigen specificity in recall antigen-specific T cells by adoptive T cell receptor transfer: implications for adoptive immunotherapy. Clin Cancer Res. 2007;13(13):4009–15. CrossRef
11. Wu T, Dai Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2017;387:61–8. CrossRef
12. Shao C, Yang F, Miao S, Liu W, Wang C, Shu Y, et al. Role of hypoxia-induced exosomes in tumor biology. Mol Cancer. 2018;17:120. CrossRef
13. Yang ZJ, Bi QC, Gan LJ, Zhang LL, Wei MJ, Hong T, et al. Exosomes derived from glioma cells under hypoxia promote angiogenesis through up-regulated exosomal connexin 43. Int J Med Sci. 2022;19(7):1205–15. CrossRef
14. Zhang W, Bai M, Liu K, Tan J, Ma J, Zhao J, et al. LncRNA surfactant associated 1 activates large tumor suppressor kinase 1/Yes-associated protein pathway via modulating hypoxic exosome-delivered miR-4766–5p to inhibit lung adenocarcinoma metastasis. Int J Biochem Cell Biol. 2022;153:106317. CrossRef
15. Chiarini F, Lonetti A, Evangelisti C, Buontempo F, Orsini E, Evangelisti C, et al. Advances in understanding the acute lymphoblastic leukemia bone marrow microenvironment: from biology to therapeutic targeting. Biochim Biophys Acta Mol Cell Res. 2016;1863(3):449–63. CrossRef
16. Milane L, Singh A, Mattheolabakis G, Suresh M, Amiji MM. Exosome mediated communication within the tumor microenvironment. J Control Release. 2015;219:278–94. CrossRef
17. Wu G, Ding X, Quan G, Xiong J, Li Q, Li Z, et al. Hypoxia-induced miR-210 promotes endothelial cell permeability and angiogenesis via exosomes in pancreatic ductal adenocarcinoma. Biochem Res Int. 2022;2022:Article ID 7752277. CrossRef
18. Mo F, Xu Y, Zhang J, Zhu L, Wang C, Chu X, et al. Effects of hypoxia and radiation-induced exosomes on migration of lung cancer cells and angiogenesis of umbilical vein endothelial cells. Radiat Res. 2020;194(1):71–80. CrossRef
19. Wigerup C, Påhlman S, Bexell D. Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol Ther. 2016;164:152–69. CrossRef
20. Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer. 2011;11:393–410. CrossRef
21. Nelson DA, Tan TT, Rabson AB, Anderson D, Degenhardt K, White E. Hypoxia and defective apoptosis drive genomic instability and tumorigenesis. Genes Dev. 2004;18:2095–107. CrossRef
22. Pilon-Thomas S, Kodumudi KN, El-Kenawi AE, Russell S, Weber AM, Luddy K, et al. Neutralization of tumor acidity improves antitumor responses to immunotherapy. Cancer Res. 2016;76(6):1381–90. CrossRef
23. Sgarbi G, Gorini G, Liuzzi F, Solaini G, Baracca A. Hypoxia and IF1 expression promote ROS decrease in cancer cells. Cells. 2018;7(7):64. CrossRef
24. Zhu X, Zuo L. Characterization of oxygen radical formation mechanism at early cardiac ischemia. Cell Death Dis. 2013;4:e787. CrossRef
25. Guzy RD, Hoyos B, Robin E, Simon MC, Hammerling U, Schumacker PT. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005;1(6):P401–8. CrossRef
26. Nita M, Grzybowski A. The role of the reactive oxygen species and oxidative stress in the pathomechanism of the age-related ocular diseases and other pathologies of the anterior and posterior eye segments in adults. Oxid Med Cell Longev. 2016;2016:Article ID 3164734. CrossRef
27. Syu JP, Chi JT, Kung HN. Nrf2 is the key to chemotherapy resistance in MCF7 breast cancer cells under hypoxia. Oncotarget. 2016;7:14659–72. CrossRef
28. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23(11):P549–55. CrossRef
29. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced: MDSC-mediated T cell activation. J Exp Med. 2014;211(5):781–90. CrossRef
30. Zub KA, de Sousa MML, Sarno A, Sharma A, Demirovic A, Rao S, et al. Modulation of cell metabolic pathways and oxidative stress signaling contribute to acquired Melphalan resistance in multiple myeloma cells. PLoS One. 2015;10(3):e0119857. CrossRef
31. Triner D, Shah YM. Hypoxia-inducible factors: a central link between inflammation and cancer. J Clin Investig. 2016;126(10):3689–98. CrossRef
32. Cummins EP, Oliver KM, Lenihan CR, Fitzpatrick SF, Bruning U, Scholz CC, et al. NF-κB Links CO2 Sensing to innate immunity and inflammation in mammalian cells. J Immunol. 2010;185(7):4439–45. CrossRef
33. De la Garza MM, Cumpian AM, Daliri S, Castro-Pando S, Umer M, Gong L, et al. COPD-Type lung inflammation promotes K-ras mutant lung cancer through epithelial HIF-1α mediated tumor angiogenesis and proliferation. Oncotarget. 2018;9:32972–83. CrossRef
34. Naldini A, Filippi I, Miglietta D, Moschetta M, Giavazzi R, Carraro F. Interleukin-1β regulates the migratory potential of MDAMB231 breast cancer cells through the hypoxia-inducible factor-1α. Eur. J Cancer. 2010;46(18):PP3400–8. CrossRef
35. Maddika S, Ande SR, Panigrahi S, Paranjothy T, Weglarczyk K, Zuse A, et al. Cell survival, cell death and cell cycle pathways are interconnected: implications for cancer therapy. Drug Resist Updat. 2007;10(1-2):13–29. CrossRef
36. Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature. 1998;391:43–50. CrossRef
37. Mow BMF, Blajeski AL, Chandra J, Kaufmann SH. Apoptosis and the response to anticancer therapy. Curr Opin Oncol. 2001;13(6):453–62. CrossRef
38. Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med. 2000;6:513–9. CrossRef
39. Erler JT, Cawthorne CJ, Williams KJ, Koritzinsky M, Wouters BG, Wilson C, et al. Hypoxia-mediated down-regulation of bid and bax in tumors occurs via hypoxia-inducible factor 1-dependent and -independent mechanisms and contributes to drug resistance. Mol Cell Biol. 2004;24(7):2875–89. CrossRef
40. Hunter FW, Young RJ, Shalev Z, Vellanki RN, Wang J, Gu Y, et al. Identification of P450 oxidoreductase as a major determinant of sensitivity to hypoxia-activated prodrugs. Cancer Res. 2015;75(19):4211–23. CrossRef
41. Cui XY, Tinholt M, Stavik B, Dahm AEA, Kanse S, Jin Y, et al. Effect of hypoxia on tissue factor pathway inhibitor expression in breast cancer. J Thromb Haemost. 2016;14(2):387–96. CrossRef
42. Jögi A, Ehinger A, Hartman L, Alkner S. Expression of HIF-1α is related to a poor prognosis and tamoxifen resistance in contralateral breast cancer. PLoS One. 2019;14(12):e0226150. CrossRef
43. Xiong G, Stewart RL, Chen J, Gao T, Scott TL, Samayoa LM, et al. Collagen prolyl 4-hydroxylase 1 is essential for HIF-1α stabilization and TNBC chemoresistance. Nat Commun. 2018;9:4456. CrossRef
44. Phillips RM, Hendriks HR, Peters GJ. EO9 (Apaziquone): from the clinic to the laboratory and back again. Br J Pharmacol. 2013;168(1):11–8. CrossRef
45. Shukla SK, Purohit V, Mehla K, Gunda V, Chaika NV, Vernucci E, et al. MUC1 and HIF-1alpha signaling crosstalk induces anabolic glucose metabolism to impart gemcitabine resistance to pancreatic cancer. Cancer Cell. 2017;32(3):392. CrossRef
46. Li CJ, Liao WT, Wu MY, Chu PY. New insights into the role of autophagy in tumor immune microenvironment. Int J Mol Sci. 2017;18(7):1566. CrossRef
47. Assi M, Kimmelman AC. Impact of context-dependent autophagy states on tumor progression. Nat Cancer. 2023;4:596–607. CrossRef
48. Singhal SK, Byun JS, Park S, Yan T, Yancey R, Caban A, et al. Kaiso (ZBTB33) subcellular partitioning functionally links LC3A/B, the tumor microenvironment, and breast cancer survival. Commun Biol. 2021;4:150. CrossRef
49. Yu S, Wang Y, Jing L, Claret FX, Li Q, Tian T, et al. Autophagy in the ‘inflammation-carcinogenesis’ pathway of liver and HCC immunotherapy. Cancer Lett. 2017;411:82–9. CrossRef
50. Meymandi ARP, Akbari B, Soltantoyeh T, Hadjati J, Klionsky DJ, Badie B, et al. Crosstalk between autophagy and metabolic regulation of (CAR) T cells: therapeutic implications. Front Immunol. 2023;14:1212695. CrossRef
51. Liu A, Li Y, Shen L, Li N, Shen L, Li Z. Pan-cancer analysis of a novel indicator of necroptosis with its application in human cancer. Aging (Albany. NY). 2022;14(18):7587–616. CrossRef
52. Xu W, Wei Q, Han M, Zhou B, Wang H, Zhang J, et al. CCL2-SQSTM1 positive feedback loop suppresses autophagy to promote chemoresistance in gastric cancer. Int J Biol Sci. 2018;14(9):1054–66. CrossRef
53. Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10(1):51–64. CrossRef
54. Rouschop KMA, van den Beucken T, Dubois L, Niessen H, Bussink J, Savelkouls K, et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Invest. 2010;120:127–41. CrossRef
55. Yang M, Zeng P, Kang R, Yu Y, Yang L, Tang D, et al. S100A8 Contributes to drug resistance by promoting autophagy in leukemia cells. PLoS One. 2014;9(5):e97242. CrossRef
56. Goncharova EA, Goncharov DA, Eszterhas A, Hunter DS, Glassberg MK, Yeung RS, et al. Tuberin regulates p70 S6 kinase activation and ribosomal protein S6 phosphorylation: a role for the TSC2 tumor suppressor gene in pulmonary lymphangioleiomyomatosis (LAM). J Biol Chem. 2002;277(34):30958–67. CrossRef
57. Kwiatkowski DJ, Zhang H, Bandura JL, Heiberger KM, Glogauer M, el-Hashemite N, et al. A mouse model of TSC1 reveals sex-dependent lethality from liver hemangiomas, and up-regulation of p70S6 kinase activity in Tsc1 null cells. Hum Mol Genet. 2002;11(5):525–34. CrossRef
58. Kenerson HL, Aicher LD, True LD, Yeung RS. Activated mammalian target of rapamycin pathway in the pathogenesis of tuberous sclerosis complex renal tumors. Cancer Res. 2002;62(20):5645–50.
59. Karbowniczek M, Yu J, Henske EP. Renal angiomyolipomas from patients with sporadic lymphangiomyomatosis contain both neoplastic and non-neoplastic vascular structures. Am J Pathol. 2003;162(2):491–500. CrossRef
60. El-Hashemite N, Zhang H, Henske EP, Kwiatkowski DJ. Mutation in TSC2 and activation of mammalian target of rapamycin signalling pathway in renal angiomyolipoma. Lancet. 2003;361(9366):1348–9. CrossRef
61. Curatolo P, Bjørnvold M, Dill PE, Ferreira JC, Feucht M, Hertzberg C, et al. The role of mTOR inhibitors in the treatment of patients with tuberous sclerosis complex: evidence-based and expert opinions. Drugs. 2016;76:551–65. CrossRef
62. Pfirmann P, Combe C, Rigothier C. Tuberous sclerosis complex: a review. Revue de Med Interne. 2021;42(10):714–21. CrossRef
63. Bar C. Delmas J, Bessou P, Morice-Picard F, Pedespan JM. Sclérose tubéreuse de Bourneville: actualités et perspectives. Perfectionnement Pédiatr. 2022;5(3):213–20.
64. Crino PB. Molecular pathogenesis of tuber formation in tuberous sclerosis complex. J Child Neurol. 2004;19:716–25. CrossRef
65. Maldonado M, Baybis M, Newman D, Kolson DL, Chen W, McKhann G, et al. Expression of ICAM-1, TNF-α, NFκB, and MAP kinase in tubers of the tuberous sclerosis complex. Neurobiol Dis. 2003;14(2):279–90. CrossRef
66. Manning BD, Logsdon MN, Lipovsky AI, Abbott D, Kwiatkowski DJ, Cantley LC. Feedback inhibition of Akt signaling limits the growth of tumors lacking Tsc2. Genes Dev. 2005;19:1773–8. CrossRef
67. Zhang H, Bajraszewski N, Wu E, Wang H, Moseman AP, Dabora SL, et al. PDGFRs are critical for PI3K/Akt activation and negatively regulated by mTOR. J Clin Invest. 2007;117:730–8. CrossRef
68. Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124(3):471–84. CrossRef
69. Yang Q, Guan KL. Expanding mTOR signaling. Cell Res. 2007;17:666–81. CrossRef
70. Corradetti MN, Guan KL. Upstream of the mammalian target of rapamycin: do all roads pass through mTOR?. Oncogene. 2006;25:6347–60. CrossRef
71. Kwiatkowski DJ, Manning BD. Tuberous sclerosis: a GAP at the crossroads of multiple signaling pathways. Hum Mol Genet. 2005;14(Issue suppl_2):R251–8. CrossRef
72. Wienecke R, König A, DeClue JE. Identification of tuberin, the tuberous sclerosis-2 product: tuberin possesses specific Rap1GAP activity. J Biol Chem. 1995;270(27):16409–14. CrossRef
73. Budanov AV, Shoshani T, Faerman A, Zelin E, Kamer I, Kalinski H, et al. Identification of a novel stress-responsive gene Hi95 involved in regulation of cell viability. Oncogene. 2002;21:6017–31. CrossRef
74. Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14(14):1296–302. CrossRef
75. Jacinto E, Loewith R, Schmidt A, Lin S, Rüegg MA, Hall A, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6:1122–8. CrossRef
76. Burnett PE, Barrow RK, Cohen NA, Snyder SH, Sabatini DM. RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc. Natl Acad Sci USA. 1998;95(4):1432–7. CrossRef
77. Pearson RB, Dennis PB, Han JW, Williamson NA, Kozma SC, Wettenhall RE, et al. The principal target of rapamycin-induced p70(s6k) inactivation is a novel phosphorylation site within a conserved hydrophobic domain. EMBO J. 1995;14:5279–87. CrossRef
78. Shimobayashi M, Hall MN. Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nat Rev Mol Cell Biol. 2014;15:155–62. CrossRef
79. Hardie DG. New roles for the LKB1→AMPK pathway. Curr Opin Cell Biol. 2005;17(2):167–73. CrossRef
80. Zheng Y, Tao Y, Zhan X, Wu Q. Nuclear receptor 4A1 (NR4A1) silencing protects hepatocyte against hypoxia-reperfusion injury in vitro by activating liver kinase B1 (LKB1)/AMP-activated protein kinase (AMPK) signaling. Bioengineered. 2022;13(4):8349–59. CrossRef
81. Bourouh M, Marignani PA. The tumor suppressor kinase LKB1: metabolic nexus. Front Cell Dev Biol. 2022;10:881297. CrossRef
82. Wang X, Meng D, Chang Q, Pan J, Zhang Z, Chen G, et al. Arsenic inhibits neurite outgrowth by inhibiting the LKB1-AMPK signaling pathway. Environ Health Perspect. 2010;118(5):627–34. CrossRef
83. Jiang ZZ, Hu MW, Ma XS, Schatten H, Fan HY, Wang ZB, et al. LKB1 acts as a critical gatekeeper of ovarian primordial follicle pool. Oncotarget. 2016;7:5738–53. CrossRef
84. Inoki K, Ouyang H, Zhu T, You M, Williams BO, Guan K-L. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. 2006;126(5):955–68. CrossRef
85. Arsham AM, Howell JJ, Simon MC. A novel hypoxia-inducible factor-independent hypoxic response regulating mammalian target of rapamycin and its targets. J Biol Chem. 2003;278(32):29655–60. CrossRef
86. Liu L, Cash TP, Jones RG, Keith B, Thompson CB, Simon MC. Hypoxia-induced energy stress regulates mRNA translation and cell growth. Mol Cell. 2006;21(4):521–31. CrossRef
87. Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, et al. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004;18:2893–904. CrossRef
88. SoferA, Lei K, Johannessen CM, Ellisen LW. Regulation of mTOR and cell growth in response to energy stress by REDD1. Mol Cell Biol. 2005;25(14):5834–45. CrossRef
89. Kirkwood JM, Tarhini AA, Panelli MC, Moschos SJ, Zarour HM, Butterfield LH, et al. Next generation of immunotherapy for melanoma. J Clin Oncol. 2008;26(20):3445–55. CrossRef
90. Hogan SA, Levesque MP, Cheng PF. Melanoma immunotherapy: next-generation biomarkers. Front Oncol. 2018;8:178. CrossRef
91. Luke JJ, Ott PA. PD-1 pathway inhibitors: the next generation of immunotherapy for advanced melanoma. Oncotarget. 2015;6:3479–92. CrossRef
92. Dhanak D, Edwards JP, Nguyen A, Tummino PJ. Small-molecule targets in immuno-oncology. Cell Chem Biol. 2017;24(9):1148–60. CrossRef
93. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–64. CrossRef
94. Sitkovsky MV, Hatfield S, Abbott R, Belikoff B, Lukashev D, Ohta A. Hostile, hypoxia-A2-adenosinergic tumor biology as the next barrier to overcome for tumor immunologists. Cancer Immunol Res. 2014;2(7):598–605. CrossRef
95. Cekic C, Linden J. Purinergic regulation of the immune system. Nat Rev Immunol. 2016;16:177–92. CrossRef
96. Vijayan D, Young A, Teng MW, Smyth MJ. Targeting immunosuppressive adenosine in cancer. Nat Rev Cancer. 2017;17(12):709–24.
97. Kumar V. Adenosine as an endogenous immunoregulator in cancer pathogenesis: where to go?. Purinergic Signal. 2013;9:145–d65. CrossRef
98. Häusler SFM, Del Barrio IM, Diessner J, Stein RG, Strohschein J, Hönig A, et al. Anti-CD39 and anti-CD73 antibodies A1 and 7G2 improve targeted therapy in ovarian cancer by blocking adenosine-dependent immune evasion Am J Transl Res. 2014;6(2):129–39.
99. Turcotte M, Spring K, Pommey S, Chouinard G, Cousineau I, George J, et al. CD73 is associated with poor prognosis in high-grade serous ovarian cancer. Cancer Res. 2015;75(21):4494–503. CrossRef
100. Stagg J, Divisekera U, Duret H, Sparwasser T, Teng MWL, Darcy PK, et al. CD73-deficient mice have increased antitumor immunity and are resistant to experimental metastasis. Cancer Res. 2011;71(8):2892–900. CrossRef
101. Ryzhov SV, Pickup MW, Chytil A, Gorska AE, Zhang Q, Owens P, et al. Role of TGF-β signaling in generation of CD39+CD73+ myeloid cells in tumors. J Immunol. 2014;193(6):3155–64. CrossRef
102. Allard B, Turcotte M, Spring K, Pommey S, Royal I, Stagg J. Anti-CD73 therapy impairs tumor angiogenesis. Int J Cancer. 2014;134(6):1466–73. CrossRef
103. Thibaudin M, Chaix M, Boidot R, Végran F, Derangère V, Limagne E, et al. Human ectonucleotidase-expressing CD25high Th17 cells accumulate in breast cancer tumors and exert immunosuppressive functions. Oncoimmunology. 2015;5(1):e1055444. CrossRef
104. Ohta A, Madasu M, Subramanian M, Kini R, Jones G, Choukèr A, et al. Hypoxia-induced and A2A adenosine receptorindependent T-cell suppression is short lived and easily reversible. Int Immunol. 2014;26(2)83–91. CrossRef
105. Beavis PA, Milenkovski N, Henderson MA, John LB, Allard B, Loi S, et al. Darcy PK. Adenosine receptor 2A blockade increases the efficacy of anti-PD-1 through enhanced antitumor T-cell responses. Cancer Immunol Res. 2015;3(5):506–17. CrossRef
106. Leclerc BG, Charlebois R, Chouinard G, Allard B, Pommey S, Saad F, et al. CD73 expression is an independent prognostic factor in prostate cancer. Clin Cancer Res. 2016;22(1):158–66. CrossRef
107. Bao R, Hou J, Li Y, Bian J, Deng X, Zhu X, et al. Adenosine promotes Foxp3 expression in Treg cells in sepsis model by activating JNK/AP-1 pathway. Am J Transl Res. 2016;8(5):2284–92.
108. Cekic C, Day YJ, Sag D, Linden J, Myeloid expression of adenosine a2A receptor suppresses T and NK cell responses in the solid tumor microenvironment, Cancer Res. 2014;74(24):7250–9. CrossRef
109. Csóka B, Zsolt Selmeczy Z, Koscsó B, Németh ZH, Pacher P, Murray PJ, et al. Adenosine promotes alternative macrophage activation via A2A and A2B receptors. FASEB J. 2012;26(1):376–86. CrossRef
110. Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MKK, et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci USA. 2006;103(35)13132–7. CrossRef
111. Iannone R, Miele L, Maiolino P, Pinto A, Morello S, Adenosine limits the therapeutic effectiveness of anti-CTLA4 mAb in a mouse melanoma model. Am J Cancer Res. 2014;4(2):172–81.
112. Vecchio EA, Tan CYR, Gregory KJ, Christopoulos A, White PJ, May LT. Ligand-independent adenosine A2B receptor constitutive activity as a promoter of prostate cancer cell proliferation, J Pharmacol Exp Ther. 2016;357(1):36–44. CrossRef
113. Cekic C, Sag D, Li Y, Theodorescu D, Strieter RM, Linden J. Adenosine A2B receptor blockade slows growth of bladder and breast tumors. J Immunol. 2012;188(1):198–205. CrossRef
114. Iannone R, Miele L, Maiolino P, Pinto A, Morello S. Blockade of A2b adenosine receptor reduces tumor growth and immune suppression mediated by myeloid-derived suppressor cells in a mouse model of melanoma. Neoplasia. 2013;15(12):1400–9. CrossRef
115. Sorrentino C, Miele L, Porta A, Pinto A, Morello S. Myeloid-derived suppressor cells contribute to A2B adenosine receptor-induced VEGF production and angiogenesis in a mouse melanoma model. Oncotarget. 2015;6:27478–89. CrossRef
116. Kjaergaard J, Hatfield S, Jones G, Ohta A, Sitkovsky M. A2A Adenosine receptor gene deletion or synthetic A2A antagonist liberate tumor-reactive CD8+ T cells from tumor-induced immunosuppression. J Immunol. 2018;201(2):782–91. CrossRef
117. Williams M, Francis J, Ghai G, Braunwalder A, Psychoyos S, Stone GA, et al. Biochemical characterization of the triazoloquinazoline, CGS 15943, a novel, non-xanthine adenosine antagonist. J Pharmacol Exp Ther. 1987;241(2):415–20.
118. Gatta F, Del Giudice M, Borioni A, Borea P, Dionisotti S, Ongini E. Synthesis of imidazo[1,2-c]pyrazolo[4,3-e]pyrimidines, pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]-pyrimidines and 1,2,4-triazolo[5,1-i]purines: new potent adenosine A2 receptor antagonists. Eur J Med Chem. 1993;28(7-8):569–76. CrossRef
119. Cekic C, Linden J. Adenosine A2A receptors intrinsically regulate CD8+ T cells in the tumor microenvironment, Cancer Res. 2014;74(24):7239–49. CrossRef
120. Ref. Deligny M, Crosignani S, Houthuys JE. Macrocyclic diamine derivatives as ent inhibitors for the treatment of cancers, and combination thereof with adenosine receptor antagonists. WO2021/20486 Patent. 2021.
121. DiRenzo D, Piovesan D, Tan J, Miles DH, Leleti MR, Park T, et al. A dual antagonist of the A2aR and A2bR adenosine receptors, relieves adenosine-mediated immune suppression. Cancer Immunol Res. 2019;7(2_Suppl.):A162. CrossRef
122. Walters MJ, Piovesan D, Tan J, DiRenzo D, Yin F, Miles D, et al. Combining adenosine receptor inhibition with AB928 and chemotherapy results in greater immune activation and tumor control. Cancer Res. 2018;78(13_Suppl):5556. CrossRef
123. Borodovsky A, Wang Y, Ye M, Shaw JC, Sachsenmeier KF, Deng N, et al. Preclinical pharmacodynamics and antitumor activity of AZD4635, a novel adenosine 2A receptor inhibitor that reverses adenosine mediated T cell suppression. Cancer Res. 2017;77(13_Suppl):5580. CrossRef