
INTRODUCTION
Tuberculosis (TB) is an infectious disease caused by the bacillus Mycobacterium tuberculosis (Mtb) that has become a serious threat for human being given the fact that it has killed approximately 1.5 million people around the world (World Health Organization, 2017). TB, especially pulmonary TB, is generally recognized for continuous three or more-week cough, fever, and severe weight loss (Ryu, 2015). Various anti-TB medications have been known, such as isoniazid, rifampicin, streptomycin, which are known as first-line medications (Alangaden et al., 1998; Cruz et al., 2018; Manabe et al., 2012; Pandey et al., 2019), in addition to amikacin, kanamycin, and capreomycin, which are called as second-line drugs (Alangaden et al., 1998; Cruz et al., 2018; Krüüner et al., 2003; Maus et al., 2005). However, issues on multi-drug resistance and extensively drug resistance of TB remain challenging (Cruz et al., 2018; Glaziou et al., 2015; Padiadpu et al., 2013). This fact highlights the urgent need to find a new chemical entity to combat the global burden of TB.
Polyketide synthase 13 (Pks13), together with the acyl-AMP ligase (FadD32), is involved in the biosynthesis of long-chain (C60-90) α-branched-β-hydroxylated fatty acid, mycolic acid, one of the main lipid components constituting thick cell wall of Mtb (Barry et al., 1998; Belardinelli and Morbidoni, 2013). Pks13 through its C-terminal thioesterase (TE) domain is involved in the key condensation step of the C40-60 meromycolate acid and saturated C26 alpha chain (Cruz et al., 2018; Portevin et al., 2004). Given the crucial role of mycolic acid to the pathology of TB, many reported that inhibition of Pks13 is a promising strategy for anti-TB therapy, as reported by Aggarwal et al. (2017), Ioerger et al. (2013), Thanna and Sucheck (2016), and Wilson et al. (2013).
Today’s era has witnessed the emerging interest in the use of virtual screening method in drug discovery processes which reduce the overall time and cost. Therefore, the present work was devoted to taking advantage of the popular method for identifying the inhibitor of Pks13 inhibitor. The virtual screening involves the use of a large compound databases for identifying small molecules potentially bind to a protein target (Kothandan et al., 2017). Virtual screening consists of ligand-based approach including pharmacophore modeling and structure-based approach including molecular docking (Kothandan et al., 2017). Ligand-based approach involves comparison of structural similarity of known and unknown compounds with an active known ligand as a query input (Dror et al., 2009), while the latter predicts the binding mode and affinity of the ligand in a drug target. This works describes the use of pharmacophore-based virtual screening, molecular docking, and molecular dynamics simulation to identify inhibitors of Pks13. The combined method is powerful in hit identification of a molecular target (Arba et al., 2018a; 2018b).
COMPUTATIONAL METHODS
Pharmacophore modeling and database screening
The ZINCPharmer web server (http://zincpharmer.csb.pitt.edu/) (Koes and Camacho, 2012) was employed for pharmacophore modeling, with the help of crystallographic structure of Polyketide synthase 13 (PDB ID: 5V3Y) and TAM16 as inhibitor. The ZINC database was employed by ZINCPharmer to perform hits compounds (Irwin et al., 2012) using a pharmacophore model of TAM16. A pharmacophore is considered as a spatial configuration of the essential features of ligand necessary for optimal binding with a specific target protein (Dror et al., 2009). The pharmacophore model was chosen for having two hydrogen bond donors, one hydrogen bond acceptor and one hydrophobic feature. The selected pharmacophore features allow to retrieve more diverse ligand of ZINC database. Each hydrogen bond donor/acceptor feature has a radius of 0.5 Å, while the hydrophobic feature has radius of 1 Å (Arba et al., 2018a).
Molecular docking and molecular dynamics simulation analysis
The obtained ligands from the virtual screening using ZINCPharmer were then subjected to molecular docking on polyketide synthase 13 (Pks13) active site employing iDock software (Li et al., 2012). The docking study is aimed to reveal the binding orientation of small molecule to a receptor protein. The crystallographic structure of Pks13 was downloaded from the Protein Data Bank with PDB ID 5V3Y (Aggarwal et al., 2017). The protein structure preparation includes adding polar hydrogen and assigning Kollman charges at physiological pH by means of AutoDockTools (Morris et al., 1998). Validation of docking protocol was achieved by redocking native ligand (TAM16) to obtain the values of root mean-square deviation (RMSD) between docked and experimental conformations.
The binding site of TAM16 was used as a center of the grid box sized 30 × 30 × 30 Å. The maximum number of binding conformations to write was 1, while other docking setting was left at default values. The Discovery Studio Visualizer (Dassault Systèmes, 2015) was employed to analysis docking results. The docked ligands were ranked based on their binding energies and four ligands having the lowest binding energies and the best conformations were subjected for further molecular dynamics simulation.
In the present study, the molecular dynamics (MD) simulations were performed on four top hit compounds, each complexed with Pks13, using the GPU version of the PMEMD engine of the Amber 16 package (Case et al., 2005; Salomon-Ferrer et al., 2013). The ff14SB (Maier et al., 2015) force field was utilized for protein, while General Amber Force Field (GAFF) (Wang et al., 2004) force fields and AM1-BCC (Jakalian et al., 2002) were used to parameterize small molecules. MD simulation was performed for 40 ns. The details of MD procedure follow our previous work (Arba et al., 2018b). Subsequent to the MD simulation, the binding free energy on 200 snapshots taken from 20–40 ns simulation trajectories was computed using the Molecular Mechanics-Poisson Boltzmann solvent accessible surface area (MM-PBSA) method of single trajectory of complex as described in our previous work (Arba et al., 2016; Kollman et al., 2000).
RESULTS AND DISCUSSION
ZINCPharmer was employed for performing virtual screening, in which several pharmacophore features were chosen, including two hydrogen bond donors, one hydrogen bond acceptor, and one hydrophobic feature (Fig. 1). Through that scheme, 107 compounds out of 22,723,923 small molecules of ZINC database were yielded.
Furthermore, the 107 compounds were docked on Pks13 active site using iDock to predict the binding conformation and affinity. It was found that the affinity of the ligands ranged from −3.92 to −12.39 kcal/mol, while the affinity of TAM16 was −10.30 kcal/mol. Out of 107 compounds, 16 compounds had lower binding energies compared to that of TAM16. Furthermore, four top scored molecules, i.e., Lig79/ZINC09281113 (E = −12.39 kcal/mol), Lig94/ZINC09584070 (E = −12.17 kcal/mol), Lig95/ZINC09209668 (E = −11.40 kcal/mol), and Lig97/ZINC09216165 (E = −12.06 kcal/mol), having lowest binding energies were proceeded for MD simulation. Figure 2 displays chemical structures of four best ligands.
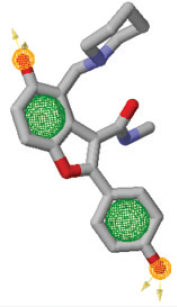 | Figure 1. The pharmacophore model of TAM16 generated by ZINCPharmer (hydrophobic: green sphere, hydrogen bond acceptor: gold spheres). [Click here to view] |
Redocking of TAM16 resulted in binding conformation with RMSD value 0.8 Å, which is acceptable for docking protocol (Morris et al., 1998). Figure 3 displays the superimposed conformations of TAM16 before docking (green) and after docking (blue).
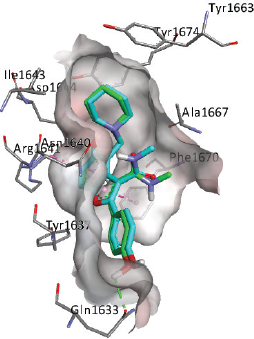
The Lig79/ZINC09281113 which exhibited the highest docking score of −11.53 kcal/mol exhibited pi-pi stacking interaction with Phe1585 and Phe1670. The same interaction occurred on other hit compounds, i.e., Lig94/ZINC09584070, Lig95/ZINC09209668, and Lig97/ZINC09216165. Besides, the pi-pi T-shaped interaction with Phe1637, amide-pi stacking interaction with Phe1585, and pi-alkyl interaction with Ala1617 and Val1614 were detected on the all four compounds. Meanwhile, additional pi-pi stacking with Tyr1582 and pi-pi T-shaped interactions with Trp1579 were observed in Lig95 interaction. Figure 4 displays the interactions of Lig79, Lig94, Lig95, and Lig97 in the polyketide synthase 13. Overall, the virtual screening using iDock showed that the interactions of four best hits to Pks13 were dominated by the hydrophobic interaction.
Molecular dynamics simulations
Each polyketide synthase 13 complexed with top four hits and TAM16 was converged during 40-ns MD simulation as depicted by the values of the RMSD and the root mean square fluctuation (RMSF). Figures 5 and 6 show RMSD and RMSF values, respectively, for each ligand-Pks13 complex during 40-ns MD simulation. It can be seen in Figure 5 that the hit compounds, i.e., Lig79, Lig94, Lig95, and Lig97 are more stable than native ligand (TAM16, red line) throughout simulation. The hit stabilities occurred since 10 ns of the simulation. The similar trend was observed in RMSF plot. The TAM16 is more fluctuated than hit compounds, as indicated by its higher RMSF values. Principally, all compounds show a similar fluctuation pattern on all amino acid residues of the protein. There were three noticeable peaks of RMSF plot, i.e., Pro117 (Pro1569), Asn139 (Asn1591), and Ala165 (Ala1617). Both Pro1569 and Asn139 (Asn1591) were loop regions which were principally more flexible than other region of protein. In addition, the highest peak at Ala165 (Ala1617) corresponded to the α-helix end.
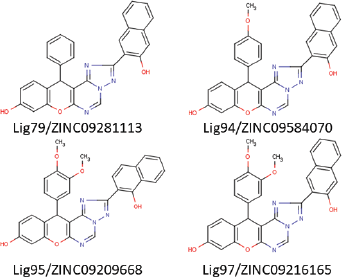 | Figure 2. The chemical structures of four best hits. [Click here to view] |
MM-PBSA binding free energy
To reveal the binding forces of each ligand, the binding free energy calculation was performed using MM-PBSA method (Table 1). Table 1 shows that the hit compounds had better affinities than that of TAM16 (Lig79: −32.00 ± 3.76 kcal/mol, Lig94: −34.96 ± 3.70 kcal/mol; Lig97: −35.16 ± 3.80 kcal/mol; Lig95: −39.24 ± 3.02 kcal/mol, 5V8: −27.36 ± 3.72 kcal/mol). Moreover, the binding affinity of Lig95 was the strongest among those of other compounds.
The favorable energy contribution was originated from the electrostatic energy (ΔEELE), van der Waals (ΔEvdw), and the nonpolar solvation energies (ΔEPBSUR) terms. On the other hand, the polar energies of desolvation (ΔEPBCAL) were unfavorable for ligand binding, which lead to unfavorable net electrostatic contributions (ΔEELE + ΔEPBCAL). The largest contribution of van der Waals energy terms clearly indicates that the binding of ligand was mainly supported by the non-covalent interaction.
 | Figure 3. Superimposed conformations of TAM16 before docking (green) and after docking (blue). The hydrogen bonds are displayed in green dashed lines. [Click here to view] |
.png) | Figure 4. The representation of docked (a) Lig79, (b) Lig94, (c) Lig95 and (d) Lig97 into binding site of polyketide synthase 13. The pink dashed line indicates pi-pi stacking, the green dashed line demonstrates pi-alkyl interactions. [Click here to view] |
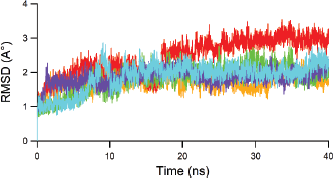 | Figure 5. The RMSD value of ligand-Pks13 complex during 40-ns dynamics runs for each 5V8 (red), Lig79 (orange), Lig94 (green), lig95 (blue) and Lig97 (purple). [Click here to view] |
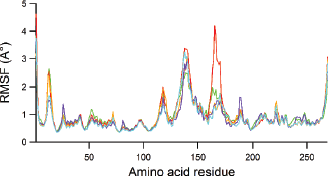 | Figure 6. The representation of RMSF plot characterizing amino acid residue fluctuation for each 5V8 (red), Lig79 (orange), Lig94 (green), lig95 (blue) and Lig97 (purple). [Click here to view] |
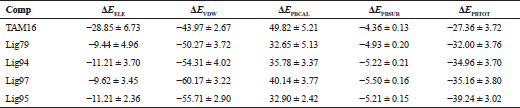 | Table 1. The binding free energy terms (kcal/mol) of each ligand bound to Pks13. [Click here to view] |
CONCLUSION
In conclusion, several hit compounds, i.e., Lig79/ZINC09281113, Lig94/ZINC09584070, Lig95/ZINC09209668, and Lig97/ZINC09216165, were acquired as potential Pks13 inhibitors by employing pharmacophore-based virtual screening, molecular docking, and molecular dynamics simulation analysis. The four hit compounds show better binding affinities compared to that of TAM16, a known inhibitor of Pks13, as evidenced by binding free energy calculation using MM-PBSA method. The binding mechanism of the hits is primarily due to van der Waals and electrostatic interactions. The present study offers new chemicals of novel Pks13 inhibitors to be evaluated in experimental study.
CONFLICT OF INTERESTS
Author declares that there are no conflicts of interest.
FINANCIAL SUPPORT AND SPONSORSHIP
None.
REFERENCES
Aggarwal A, Parai MK, Shetty N, Wallis D, Woolhiser L, Hastings C, Dutta NK, Galaviz S, Shakal RC, Shrestha R, Wakabayashi S, Walpole C, Matthews D, Floyd D, Scullion P, Riley J, Epemolu O, Norval S, Sacchettini JC. Development of a novel lead that targets M. tuberculosis polyketide synthase 13. Cell, 2017; 170(2):249–59. CrossRef
Alangaden GJ, Kreiswirth BN, Aouad A, Khetarpal M, Igno FR, Moghazeh SL, Manavathu EK, Lerner SA. Mechanism of resistance to amikacin and kanamycin in mycobacterium tuberculosis. Antimicrob Agents Ch, 1998; 42(5):1295–7. CrossRef
Arba M, Nur-Hidayat A, Surantaadmaja SI, Tjahjono DH. Pharmacophore-based virtual screening for identifying β5 subunit inhibitor of 20S proteasome. Comput Biol Chem, 2018a; 77:64–71. CrossRef
Arba M, Ihsan S, Tjahjono DH. Computational approach toward targeting the interaction of porphyrin derivatives with Bcl-2. J Appl Pharm Sci, 2018b; 8(12):60–6. CrossRef
Arba M, Kartasasmita RE, Tjahjono DH. Molecular docking and dynamics simulations on the interaction of cationic porphyrin–anthraquinone hybrids with DNA G-quadruplexes, J Biomol Struct Dyn, 2016; 34(2):427–38. CrossRef
Barry CE, Lee RE, Mdluli K, Sampson AE, Schroeder BG, Slayden RA, Yuan Y. Mycolic acids: structure, biosynthesis and physiological functions. Prog Lipid Res, 1998; 37(2):143–79. CrossRef
Belardinelli JM, Morbidoni HR. Recycling and refurbishing old antitubercular drugs: the encouraging case of inhibitors of mycolic acid biosynthesis. Expert Rev Anti Infe, 2013; 11(4):429–40. CrossRef
Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, Woods RJ. The Amber biomolecular simulation programs. J Comput Chem, 2005; 26(16):1668–88. CrossRef
Cruz JN, Costa JFS, Khayat AS, Kuca K, Barros CAL, Neto AMJC. Molecular dynamics simulation and binding free energy studies of novel leads belonging to the benzofuran class inhibitors of Mycobacterium tuberculosis Polyketide Synthase 13. J Biomol Struct Dyn, 2018; 1–12. CrossRef
Dror O, Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ. Novel approach for efficient pharmacophore-based virtual screening: method and applications. J Chem Inf Mod, 2009; 49(10):2333–43. CrossRef
Glaziou P, Sismanidis C, Floyd K, Raviglione M. Global epidemiology of tuberculosis. Csh Perspect Med, 2015; 5(2):a017798. CrossRef
Ioerger TR, O’Malley T, Liao R, Guinn KM, Hickey MJ, Mohaideen N, Murphy KC, Boshoff HIM, Mizrahi V, Rubin EJ, Sassetti CM, Barry III CE, Sherman DR, Parish T, Sacchettini JC. Identification of new drug targets and resistance mechanisms in mycobacterium tuberculosis. PLoS One, 2013; 8(9):e75245. CrossRef
Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, Coleman RG. ZINC: a free tool to discover chemistry for biology. J Chem Inf Model, 2012; 52(7):1757–68. CrossRef
Jakalian A, Jack DB, Bayly CI. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J Comput Chem, 2002; 23(16):1623–41. CrossRef
Koes DR, Camacho CJ. ZINCPharmer: pharmacophore search of the ZINC database. Nucleic Acids Res, 2012; 40(W1):W409–14. CrossRef
Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong, L, Lee M, Lee T, Duan Y, Wang W, Donini O, Cieplak P, Srinivasan J, Case DA, Cheatham TE. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res, 2000; 33(12):889–97. CrossRef
Kothandan S, Sasikala RP, Meena KS. Structure based pharmacophore modeling, virtual screening and molecular docking of potential phytochemicals against HSP70. J Appl Pharm Sci, 2017; 7(02):137–41.
Krüüner A, Jureen P, Levina K, Ghebremichael S, Hoffner S. Discordant resistance to kanamycin and amikacin in drug-resistant mycobacterium tuberculosis; Antimicrob Agents Ch, 2003; 47(9):2971–3. CrossRef
Li H, Leung K-S, Wong M-H. idock: a multithreaded virtual screening tool for flexible ligand docking. Proceedings of the 2012 IEEE Symposium on Computational Intelligence in Bioinformatics and Computational Biology (CIBCB), pp. 77–84, 2012. CrossRef
Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput, 2015; 11(8):3696–713. CrossRef
Manabe YC, Hermans SM, Lamorde M, Castelnuovo B, Mullins CD, Kuznik A. Rifampicin for continuation phase tuberculosis treatment in Uganda: a cost-effectiveness analysis. PLoS One, 2012; 7(6):e39187. CrossRef
Maus CE, Plikaytis BB, Shinnick TM. Molecular analysis of cross-resistance to capreomycin, kanamycin, amikacin, and viomycin in mycobacterium tuberculosis. Antimicrob Agents Ch, 2005; 49(8):3192–7. CrossRef
Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem, 1998; 19(14):1639–62. CrossRef
Padiadpu J, Mukherjee S, Chandra N. Rationalization and prediction of drug resistant mutations in targets for clinical anti-tubercular drugs. J Biomol Struct Dyn, 2013; 31(1):44–58. CrossRef
Pandey B, Grover S, Goyal S, Jamal S, Singh A, Kaur J, Grover A. Novel missense mutations in gidB gene associated with streptomycin resistance in Mycobacterium tuberculosis: insights from molecular dynamics. J Biomol Struct Dyn, 2019; 37(1):20–35. CrossRef
Portevin D, de Sousa-D-D'Auria C, Houssin C, Grimaldi C, Chami M, Daffé M, Guilhot C. A polyketide synthase catalyzes the last condensation step of mycolic acid biosynthesis in mycobacteria and related organisms. Proc Natl Acad Sci USA, 2004; 101(1):314–9. CrossRef
Ryu YJ. Diagnosis of pulmonary tuberculosis: recent advances and diagnostic algorithms. Tuberc Respir Dis, 2015; 78(2):64–71. CrossRef
Salomon-Ferrer R, Götz AW, Poole D, Le Grand S, Walker RC. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh ewald. J Chem Theory Comput, 2013; 9(9):3878–88. CrossRef
Thanna S, Sucheck SJ. Targeting the trehalose utilization pathways of Mycobacterium tuberculosis. Med Chem Commun, 2016; 7(1):69–85. CrossRef
Wang JM, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem, 2004; 25(9):1157–74. CrossRef
Wilson R, Kumar P, Parashar V, Vilchèze C, Veyron-Churlet R, Freundlich JS, Barnes SW, Walker JR, Szymonifka MJ, Marchiano E, Shenai S, Colangeli R, Jacobs Jr WR, Neiditch MB, Kremer L, Alland D. Antituberculosis thiophenes define a requirement for Pks13 in mycolic acid biosynthesis. Nat Chem Biol, 2013; 9:499–506. CrossRef
World Health Organization. Global tuberculosis report 2017. Available via http://www.who.int/tb/publications/global_report/en/