
INTRODUCTION
Dengue fever is an arboviral infection that has turned to be a major health concern globally in the recent years. Reports state that 100 million cases of dengue fever and 500,000 cases of dengue hemorrhagic fever occur annually, mainly in tropical countries, especially in Asia, Latin America, and the Caribbean (Deen et al., 2006; Mackenzie et al., 2004). Dengue virus, the etiological agent causing dengue infection, has four distinct serotypes (DENV-1, DENV-2, DENV-3, and DENV-4) belonging to the genus Flavivirus. The genome of dengue virus is a single stranded, positive-sense RNA virus with 11 kb in length. The genome of dengue virus encodes for three structural and seven non-structural proteins responsible for both viral and cellular functions (Lindenbach et al., 2001). The dengue infection is initiated by the fusion of viral membrane on the host cell membrane that is mediated by one of the potential proteins, the E protein in a pH dependent manner (Stiasny et al., 2006). The E protein consists of three functional domains central domain I, extended finger like domain II, and immunoglobulin-like domain III (Modis et al., 2003; 2004; 2005; Zhang et al., 2004).
Such similar three domain organizations are seen in other virus of this family, the tick-borne encephalitis virus (Rey et al., 1995) , West Nile virus (Kanai et al., 2006; Nybakken et al., 2006), and the E1 protein of Semliki Forest virus (Lescar et al., 2001). The crystal structure analyses for the E protein of dengue virus revealed a hydrophobic pocket region bound to a detergent molecule (_β-N-octylglucoside [_β-OG]), present at the hinge region between domains I and II of the E protein, an important component for the pH-triggered conformational rearrangement required for the fusion process, a significant step in invasion of the virus (Noha et al., 2011). The E protein is crucial for the fusion activity and making it a valuable target for antiviral studies.
Pharmacophore modeling has been considered as one of the successful tools in drug discovery and design (Seidel et al., 2010; Wolber et al., 2006) that provides fruitful information about the interactions between a target protein and a ligand. Three-dimensional pharmacophore modeling is based on the three-dimensional arrangement of chemical features (such as hydrogen bonds donors/acceptors, hydrophobicity, charges, and lipophilic areas) that explains the binding of a ligand to the macromolecule. The spatial arrangement of chemical features represents the essential interactions between ligands and proteins (Noha et al., 2012). Since, the structure-based pharmacophore model maintains an adequate geometric relationship of the active sites and is less dependent on the bonded inhibitor compared with the ligand-based pharmacophore model, it is expected to improve the novelty of the hit in the virtual screening.
The envelope protein is one of the potential targets and many antiviral studies have been carried out targeting the E protein and a structure based pharmacophore screening is an attempt to find the efficient compounds with the understanding of the active site conformation and the chemical features in the three-dimensional space. The bounded detergent molecule into the hydrophobic space will help in understating the chemical features attached to the pharmacophore features. This study is based on the understanding of the pharmacophore feature of the bonded β-OG within the hydrophobic pocket.
MATERIALS AND METHODS
Hits screened and enzyme crystallographic structure
In this study, compounds from different databases and phytocompounds from different sources were involved. All the used compounds were subjected into ligand preparation protocol before analyzing. The crystal structure of E protein of dengue virus [protein data bank (PDB) ID: 1OKE] complexed with β-OG molecule—a detergent molecule was obtained from the PDB. The bonded ligand molecule was used to predict the involved pharmacophore feature for the study.
Construction of structure based pharmacophore model
Structure based pharmacophore model (SBPM) was successfully used to design novel compounds with potent biological activity in many therapeutic areas (Thangapandian et al., 2011). In the present study, SBPM was generated based on the co-crystal structure of dengue virus E protein complexed with a detergent molecule β-OG (1OKE) using the receptor-ligand pharmacophore generation module of the Discovery Studio Version 4.0 (DS V 4.0, Accelrys Inc., San Diego, CA). The site sphere for the analysis was defined as 12 AËš radius that covers all the significant and important residues with the bounded inhibitor β-OG located in the hydrophobic pocket of the E protein. This process helps in the generation of SBPM, a special protocol specifically designed to determine the protein–ligand interaction at the hydrophobic pocket that creates the corresponding pharmacophoric features that includes hydrogen bond acceptor (HBA), hydrogen bond donor (HBD), and hydrophobic (HY). In addition, the excluded volumes were considered in the pharmacophore model generation to improve the effectiveness of virtual screening (Greenidge et al., 1998; Norinder, 2000; Palomer et al., 2002). A total of 10 hypothesis models are generated and out of which the best one is ranked a Hypo_1.
Screening of structure based pharmacophore model
The hypothesis model generated by the SBPM was further used as a parameter to screen effective hits from different in house and other phytocompounds isolated from various sources. The specific hydrophobe was chosen by using the cluster analysis scenario, the HBDs and HBA features were clustered, specific hydrophobe features were selected, and S interaction grouping was done keeping required feature as true, feature group true, the pharmacophore feature as 8 and subset is set as 250 and all the rest of the parameters were maintained as default. The pharmacophore model was validated by pharmprint frequency and the best compounds were selected based on the highest fit value.
Validation using molecular docking
The best compounds which fits to the obtained pharmacophore model was further validated by performing flexible docking using Discovery Studio Version 4.0 (DS V 4.0, Accelrys Inc., San Diego, CA). Before performing molecular docking, the receptor protein (1OKE) was prepared using protein preparation. The protein was first cleaned and the unwanted crystal structure, water molecules, and other bounded ligands were removed from the structure. The quality of the receptor protein was checked followed by loop refinement, and the receptor protein was validated for the presence of disallowed regions. The protein energy minimization was performed using CHARMm force field in DS. The active site used in this study has Glu49, Ala50, Leu207, Ile270, Gln271, and Thr280. All the compounds screened using the best pharmacophore feature were validated using the flexible docking.
ADMET analysis
Molecular properties of the compounds were examined using Discovery Studio 4.0. ADMET studies and the following parameters: atom based Log P98 (A LogP 98), polar surface area (PSA), blood brain barrier, Cytochrome P450, and hepatotoxicity were assessed.
RESULTS AND DISCUSSION
Structure-based pharmacophore model
Out of the 10 hypothesis pharmacophore models generated by the software with the available information of the bounded ligand (a detergent molecule β-OG within the hydrophobic pocket of the dengue virus E protein), the best one was chosen based on the fit value and number of pharmacophore features generated. The pharmacophore was generated based on the interacting amino acid residues in the hydrophobic pocket. A report suggest that this hydrophobic pocket, occupied by a detergent molecule (_-N-octylglucoside [β-OG]) lying at a hinge region between domains I and II of the E protein, is important for the low-pH-triggered conformational rearrangement required for fusion, and this pocket act as a potential site for small-molecule fusion inhibitors (Modis et al., 2003). The pharmacophore feature finally designed based on the interaction profile of the β-OG with the E protein showed the following features that includes HBAs (A1, A5, A10, A12, A14, A52, and A64) HBDs (D2, D16, D21, D30, D40, D58, and D74), hydrophobe (H8, H14, and H29). These features contribute to the chemical pharmacophore features (Fig. 1)
Screening of structure-based pharmacophore model
The mapped features were used as an input to screen the effective ligands that fits to the predicted pharmacophore features (Fig. 1). The screenings of various inbuilt and other added databases were included in this screening process. All the possible hits were generated and screening was done based on the fit value and the total number of features were matched. Maximum hits were obtained for four features with a fit value of 3. A total of 5,000 compounds were screened and obtained best 50 compounds with the highest fit value.
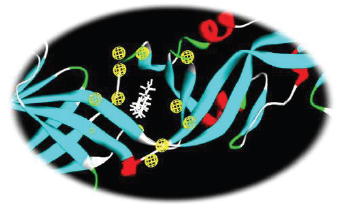 | Figure 1. Three-dimensional structure of the pharmacophore model of the E protein of dengue virus in complex with the bounded detergent molecule β-OG. [Click here to view] |
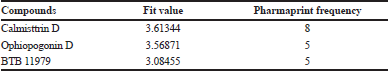
The arrangement of the chemical feature markers in the three-dimensional space was used to search molecules that fits the pharmacophore, was used as a successful approach in screening and would lead to the prediction of 15 novel compounds. The features were selected based on the ligand attached to the hydrophobic site in the E protein. A total of three hydrophobes were predicted in the pharmacophore feature out of which the compounds were screened using the hydrophobe feature 14. Maximum number of compounds were predicted with 4-feature pharmacophore. Out of the 50 compounds screened, the best three compounds and its respective details are seen in Table 1 and Figure 2.
Molecular docking
Flexible docking was done as a step to validate the screened compounds based on the pharamocophore features for the E protein complexed with a detergent molecule β-OG. The flexible docking was performed using the Discovery Studio Version 4.0 with all the parameters set keeping both the ligand and receptor in flexible form. Out of the 50 molecules incorporated for molecular docking, eight compounds exhibited binding energy against the hydrophobic pocket site of the E protein. The active site residues for the docking study were Glu49, Ala50, Leu207, Ile270, Gln271, and Thr280. The best three compounds were selected from the hits based on the binding energy. The compound Ophiopogonin D showed the highest binding energy of −146.36 Kcal/mol forming hydrogen bond with Ala50, Glu 126, Lys 128, Asn124, Lys202, Thr48, and Asp203. Similarly, the compound Calmisttrin D exhibited a binding energy of −118.735 Kcal/mol and extends hydrogen bonding with Gln271, Ala50, and Asp203. The compound BTB 11979 shows a binding energy of 99.9672 Kcal/mol which extends hydrogen bonding with Ala50, Gln 200, Gln 271, and Phe 193. The detailed analysis of the screened compounds is given in Table 2 and Figure 3.
 | Table 1. The pharmacophore features of 1OKE complexed with β-OG. [Click here to view] |
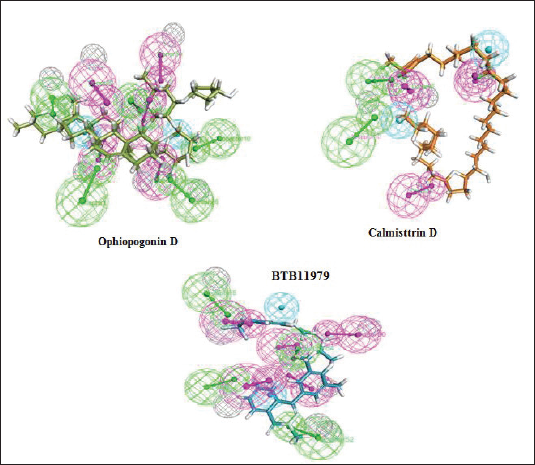 | Figure 2. The mapped pharmacophore features for the best three screened molecules were green color represent the HBAs, magenta color represents HBDs, and cyan color represents the hydrophobe feature. [Click here to view] |
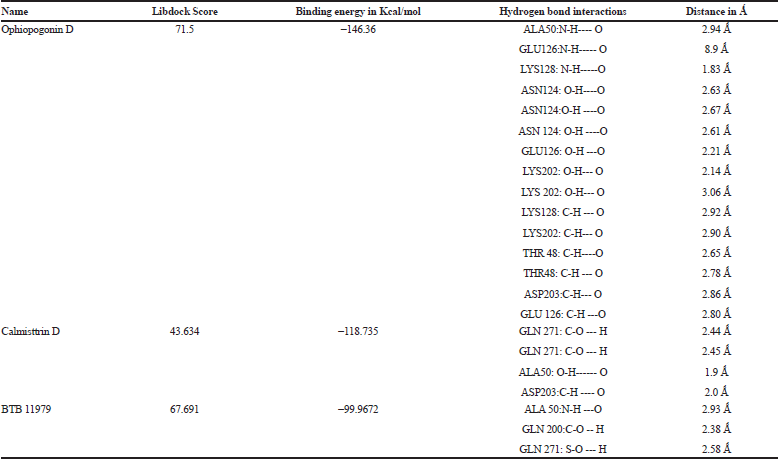 | Table 2. The interaction profile of the lead molecules against E protein of dengue virus. [Click here to view] |
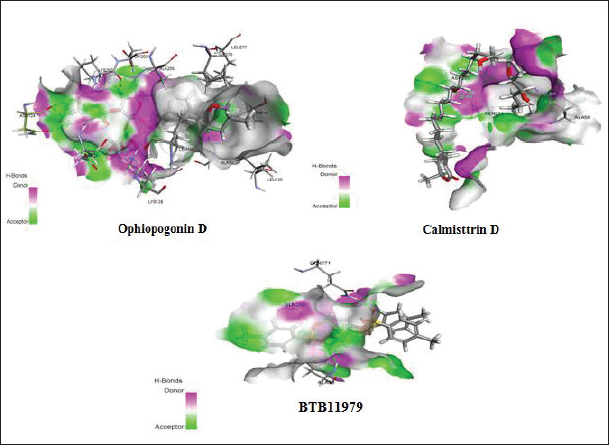 | Figure 3. Attractive hydrogen bonding interaction profile of the lead molecules within the hydrophobic pocket of dengue virus E protein. [Click here to view] |
CONCLUSION
In this study, three lead molecules namely Ophiopogonin D, Calmisttrin D, and BTB 11979 were identified based on the structure-based pharmacophore model and were validated using the flexible docking study against the E protein of dengue virus. All the three compounds were found to be non-toxic, non-carcinogenic, and non-hepatotoxic, but the compound Calmisttrin D had the highest fit value and posses’ good blood barrier permeability and high intestinal adsorption rate by ADMET analysis. It is proposed that Calmisttrin D could be a potential inhibitor of DENV E fusion protein and further in vitro and in vivo evaluation studies are suggested.
ACKNOWLEDGMENTS
This work was supported by Bioinformatics Infrastructure Facility Centre of Department of Biotechnology, Ministry of Science and Technology, Govt. of India vide Grant No.BT/BI/25/068/2012/2015.
REFERENCES
Deen JLE, Harris B, Wills A, Balmaseda SN, Hammond C, Rocha NM, Dung NT, Hung Hien TT, Farrar JJ. The WHO dengue classification and case definitions: time for a reassessment. Lancet, 2006; 368:170–3. CrossRef
Greenidge PA, Carlsson B, Bladh LG, Gillner M. Pharmacophores incorporating numerous excluded volumes defined by X-ray crystallographic structure in three-dimensional database searching: application to the thyroid hormone receptor. J Med Chem, 1998; 41:2503–12. CrossRef
Kanai R, Kar K, Anthony K, Gould LH, Ledizet M, Fikrig E, Marasco WA, Koski RA, Modis Y. Crystal structure of West Nile virus envelope glycoprotein reveals viral surface epitopes. J Virol, 2006; 80:11000–8. CrossRef
Lescar J, Roussel A, Wien MW, Navaza J, Fuller SD, Wengler G, Wengler G, Rey FA. The fusion glycoprotein shell of Semliki Forest virus: an icosahedral assembly primed for fusogenic activation at endosomal pH. Cell, 2001;105:137–48. CrossRef
Lindenbach BD, Rice CM, Knipe DM, Howley PM. Fields virology. 4th edition, Lippincott Williams & Wilkins, Philadelphia, PA, pp 991–1041, 2001.
Mackenzie JS, Gubler DJ, Petersen LR. Emerging flaviviruses: the spread and resurgence of Japanese encephalitis, West Nile and dengue viruses. Nat Med, 2004; 10:98–109. CrossRef
Modis Y, Ogata S, Clements D, Harrison SC. A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc Natl Acad Sci USA, 2003; 100:6986–91. CrossRef
Modis Y, Ogata S, Clements D, Harrison SC. Structure of the dengue virus envelope protein after membrane fusion. Nature, 2004; 427:313–9. CrossRef
Modis Y, Ogata S, Clements D, Harrison SC.Variable surface epitopes in the crystal structure of dengue virus type 3 envelope glycoprotein. J. Virol, 2005; 79:1223–31. CrossRef
Noha SM, Atanasov AG, Schuster D, Markt P, Fakhrudin N, Heiss EH, Schrammel O, Rollinger JM, Stuppner H, Dirsch VM, Wolber G. Discovery of a novel IKK-β inhibitor by ligand-based virtual screening techniques. Bioorg Med Chem Lett, 2011; 21:577–83. CrossRef
Noha SM, Jazzar B, Kuehnl S, Rollinger JM, Stuppner H, Schaible AM, Werz O, Wolber G, Schuster D. Pharmacophore-based discovery of a novel cytosolic phospholipase A2α inhibitor. Bioorg Med Chem Lett, 2012; 22:1202–7. CrossRef
Norinder U. Refinement of catalyst hypotheses using simplex optimisation. J Comput Aided Mol Des, 2000; 14: 545–57. CrossRef
Nybakken GE, Nelson CA, Chen BR, Diamond MS, Fremont DH. Crystal structure of the West Nile virus envelope glycoprotein. J Virol, 2006; 80:11467–74. CrossRef
Palomer A, Cabré F, Pascual J, Campos J, Trujillo MA, Entrena A, Gallo MA, García L, Mauleón D, Espinosa A. Identification of novel cyclooxygenase-2 selective inhibitors using pharmacophore models. J Med Chem, 2002; 45:1402–11. CrossRef
Rey FA, Heinz FX, Mandl C, Kunz C, Harrison SC. The envelope glycoprotein from tick-borne encephalitis virus at 2 Å resolution. Nature, 1995; 375:291–8. CrossRef
Seidel T, Ibis G, Bendix F, Wolber G. Strategies for 3D pharmacophore-based virtual screening. Drug Discov Today Technol, 2010; 7:e221–8. CrossRef
Stiasny K, Heinz FX. Flavivirus membrane fusion. J Gen Virol, 2006; 87:2755–66. CrossRef
Thangapandian S, John S, Sakkiah S, Lee KW. Ligand and structure based pharmacophore modeling to facilitate novel histone deacetylase 8 inhibitor design. Eur J Med Chem, 2011; 45:4409–17. CrossRef
Wolber G, Dornhofer AA, Langer T. Efficient overlay of small organic molecules using 3D pharmacophores. J Comput Aid Mol Des, 2006; 20:773–88. CrossRef
Zhang Y, Zhang W, Ogata S, Clements D, Strauss JH, Baker TS, Kuhn RJ, Rossmann MG. Conformational changes of the flavivirus E glycoprotein. Structure, 2004; 12:1607–18. CrossRef