
INTRODUCTION
Nowadays and regarding the actual world economic conjuncture on the science community, the inefficiencies of the drug discovery and development process are a forthcoming subject (Antman et al., 2012). Despite critical discoveries in science and technology, expressions such as “Valley of Death” keeps appearing to describe these inefficiencies, and so there is an urge for a shift on the business and scientific background of drug research & development (R&D) (Abou-Gharbia and Childers, 2014).
In oncology, the R&D seems to be even more complex as the success rate of drug candidates is abysmally lower when compared with the other areas of healthcare (Bhattacharjee, 2012; Williams, 2015). Besides this “curse” in R&D, developing new drugs against a cancer such as glioblastoma multiforme (GBM) is particularly challenging due to a variety of reasons related with both compound and tumor characteristics, resulting in unsuccessful approaches and drugs, culminating in a fatal outcome (Olar and Aldape, 2014). GBM is classified by the World Health Organization as a grade VI malignant astrocytoma (Schonberg et al., 2013), being the most common intrinsic primary brain tumor in adults, and represents over 80% of diffuse gliomas (Toda, 2013). In the case of this type of tumor, among other facts, their origin, structure, biology, cellular metabolism, and microenvironment are still unclear (Behnan et al., 2013; Lima et al., 2012).
One of the proteins, which is involved in many processes related to GBM, is the 3-phosphoinositide-dependent protein kinase-1 (PDK1). It acts as a master upstream protein kinase, phosphorylating and activating a subgroup of the AGC (protein kinase A, protein kinase G, protein kinase C) kinase family (which it is part of) implicated in the control of cell growth, proliferation, survival, and metabolism regulation (Nesi, 2011; Sephton et al., 2009).
In GBM, PDK1 is directly and indirectly part of critical molecular pathways, which regulate several growth factors and oncogenes, including phosphatidylinositol-3-kinase (PI3K)/PDK1/protein kinase B (Akt) pathway, PI3K–mammalian target of rapamycin (mTOR) crosstalk pathway, and phosphatase and tensin homolog (PTEN)-Akt-mTOR signaling pathway (Dunn et al., 2012; Krakstad and Chekenya, 2010). In addition, cytokines such as interleukin-6 have been reported to influence the PI3K/AKT signaling pathway (Guven-Maiorov et al., 2014). Therefore and regarding all said, it is very important to clarify the effects of inhibiting this protein & its potential as future therapeutic agent. To do so, the induced drug perturbation in glioblastoma cancer cells through in vitro assays by performing a multiparametric characterization of dose-response of two PDK1 inhibitors (G51 and FC100) was parameterized.
As there is a need to develop and improve in vitro models for drug discovery in GBM (Nesi, 2011), the overall context of this perturbation studies is the new-age pharmacology so-called quantitative and systems pharmacology (Sephton et al., 2009).
MATERIALS AND METHODS
Reagents and cells
The U-87 MG human glioblastoma astrocytoma cell line was kindly made available by the Center for Neuroscience and Cell Biology of the University of Coimbra (Portugal).
The PDK1 inhibitors called G51 (C25H25N5O4) and FC100 (C13H12N4O3S) were synthesized and provided within a collaboration with Prof. Simona Rapposelli from the Department of Pharmacy of the University of Pisa (Italy).
High glucose Dulbecco’s modified Eagle’s medium (HG-DMEM), dimethyl sulfoxide (DMSO), phosphate-buffered saline (PBS), methanol (MeOH), and sulphorhodamine B sodium salt (SRB) were purchased from Sigma-Aldrich Chemical S.L. (Sintra, Portugal). Heat-inactivated fetal bovine serum (FBS) and penicillin-streptomycin were obtained from Invitrogen (Barcelona, Spain). Tris(hydroxymethyl)aminomethane was obtained from National Diagnostics (Hessle Hull, UK).
Cell culture and drug treatment
Cells were seeded at a 1.5 × 105 cells/ml density and were grown at 37 °C under humidified atmosphere, containing 5% CO2, in HG-DMEM (4.5 g/l glucose) supplemented with 10% heat-inactivated FBS, 100 U/mol penicillin, and 100 µg/ml streptomycin.
The PDK1 inhibitors G51 and FC100 were solubilized in pure DMSO as 10 mM solutions, followed by a dilution with PBS to obtain 1 mM samples containing 10% DMSO.
For dose-response experiments, cells were plated in triplicate at a 5.7 × 104 cells/ml density in 24-well plates, allowed to adhere overnight, treated with five different dosages (2.5, 5, 10, 20, and 40 µM) of each compound, and incubated for time periods of 24, 48, and 72 hours.
In order to determine whether the PDK1 inhibitors effects were cytostatic or cytotoxic, two sets of U-87 MG cells were exposed to the compounds under the same conditions and for the same period of time. At the end of a 72 hours exposure, one set of cells was assayed while for the other the drug-containing medium was replaced by fresh culture medium. The incubation continued for an additional 72 hours, after which the plate was assayed as to cytostatic or cytotoxic effects through methods reported in the literature (Jenkins, 2013).
Measurement of drug response
The SRB assay for evaluation of cell density was used (Monks et al., 1991). Cells were fixed by adding 1 ml of cold 1% acetic acid/MeOH, followed by storage overnight at 4°C. The fixed cells were then placed at room temperature for 24 hours; 500 µl of SRB (0.5% w/v) were added to each well and they were incubated at 37°C for 1 hour. The plates were washed with 1% acetic acid to remove unbound SRB and allowed to dry overnight. SRB was solubilized with 1 ml of 10 mM Tris (pH 10) per well, shaken, and the supernatant was transferred to a 96-well plate to measure the optical density at 540 nm (on a Bio-Tek µQuant MQX200 Spectrophotometer UV-VIS).
Dose-response curve fitting and data calculations
Triplicate dose-response data were fitted to a logistical sigmoidal model (Eq. 1, where y is a response measure at dose D), using nonlinear least-squares regression (in GraphPad Prism Software, Version 5.00 for Windows).
The different dose-response parameters for each individual curve were estimated, including GI50, total growth inhibition (TGI), HS, minimum effect (Einf), and Emax or E0. Based on the type of model used to predict these parameters, a HS of −1 was considered as standard, a HS lower than −1 is steeper, and a value higher than −1 is shallow (Jenkins, 2013). In addition, the parameter area under the curve (AUC) was calculated, defined as the sum of measured responses (relative viability) at all tested drug concentrations. Hence, AUC = 9 corresponds to an inactive compound, whereas smaller AUC values represent biologically significant drug activities regarding inhibition of cell proliferation and/or cell death promotion. The drug concentration resulting in TGI was determined when the amount of protein at the end of each drug incubation period was found to be unchanged relative to the control.
The values for IC50 and % inhibition direct to PDK1 (for a 10 µM concentration) were previously obtained for the inhibitors under study at Prof. Rapposelli´s laboratory (Invitrogen Z’-LYTE® biochemical assay).
Regarding the SRB assay (Fallahi-Sichani et al., 2013), three types of measurements were performed: growth control (C0), control of treatment for each chosen time-point (C24h, C48h, and C72h), and cell population density upon treatment (T). The percentage of SRB retention was computed in GraphPad Prism Software (Version 5.00 for Windows), using the one-way analysis of variance method followed by multiple comparison Turkey’s test. Differences were considered statistically significant for p < 0.05, p < 0.01, and p < 0.001.
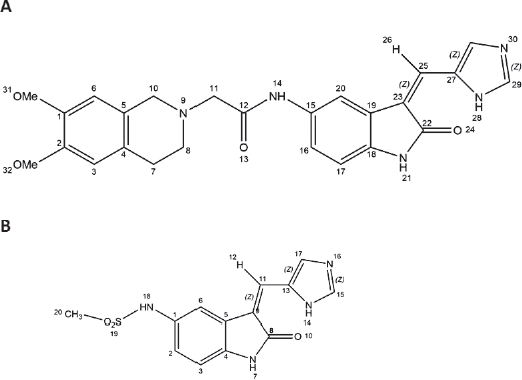 | Figure 1. Molecular structures of G51 (A) and FC100 (B) compounds. [Click here to view] |
All data were processed under high-quality control: the analysis of average, amplitude of the sample, standard deviation (SD), coefficient of variation, standard error of the mean (SEM), and Dixon’s Q test (90 % of confidence) were calculated. After computational model fitting, the goodness of fit was also evaluated by the coefficient of determination (R2).
RESULTS AND DISCUSSION
Drug-induced perturbation
The perturbation at the cellular system U-87 MG cells, by a range of dosages of two PDK1 inhibitors G51 and FC100 (Fig. 1A and B, respectively), was evaluated by SRB assay.
For G51, the most significant perturbation was verified after 72 hours of incubation for the higher dose—40 µM (Fig. 2A).
Also, the cells display a very significant perturbation at 20 µM. At 24 and 48 hours, there is no significant meaning of system perturbation. Further analysis of the systematic perturbation of the tested inhibitors on the cell response (Fig. 3) shows that when comparing the lower concentrations at 72 hours to doses in the range between 10 and 40 µM at the lower time points of treatment, there was a higher effect for the latter. In addition, for 72 hours of incubation, there were no significant differences between the 72 hours control (C72h) and the 2.5, 5, and 10 µM doses tested.
In the case of FC100, the significances marked in Figure 1B are related with the cells growth as the increase of % SRB retention across time-points in the presence of drug at the five concentration levels. These are directly correlated with the % SRB retention of control growth and as a result, there is no significant disruption on the cellular system made by this drug.
Drug perturbation effects evaluation
Analyzing the effects of induced drug perturbation, G51 drug appears to have a cytostatic effect as the cells grow and undergo mitosis 3 days after removal of the drug (time-point 144 hours), with an extremely significant mean for the 20 and 40 µM doses (Fig. 3).
Regarding the FC100 drug, the perturbation of the cellular system U-87 MG fails to produce a specific cytostatic or cytotoxic effect, irrespective of the concentration tested. In fact, even showing a significant difference between cells treated at 72 hours and cells counted 3 days after FC100 removal (time-point 144 hours), which could indicate a cytostatic effect, there is no significant reduction in the percentage of SRB retention of these cells, when compared to the control, for any of the time-points considered in the study (24, 48, and 72 hours) as shown in Figure 2B. However, by multiple comparison analysis (Fig. 4), the higher significant mean difference comparing the time-points 72 and 144 hours was for the dose 40 µM, whereas, it was not too much different from the effect of the other doses (SD = 0.07). In addition, there is no evidence of a cytotoxic effect induced by perturbation. However, FC100 was found to induce a higher cellular perturbations in the first 24 hours of treatment when compared to 48 and 72 hours (Fig. 3). It is also interesting to note that there are no significant perturbation differences between the time-points 48 and 72 hours upon treatment with the drug. This might indicate that the almost negligible perturbation due to FC100 is induced immediately—shown at 24 hours—on this cellular system. Inclusively, during the first 24 hours, the cell response to FC100 seems to be similar for all the dosages tested (SD = 0.06).
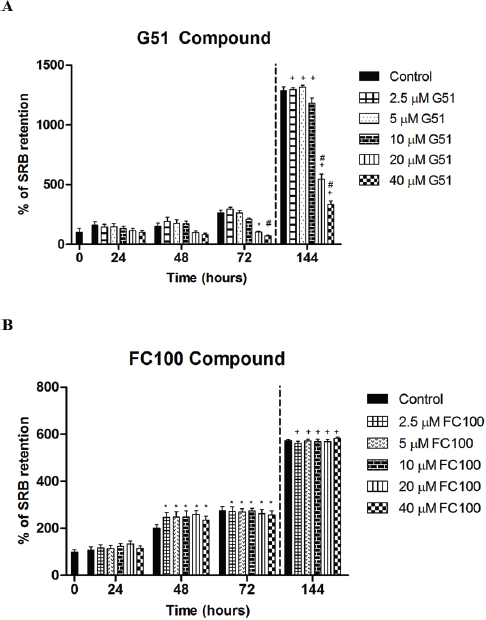 | Figure 2. Perturbation effect of G51 (A) and FC100 (B) compounds at the cellular system-level of U87-MG cell line, n = 3. The bars represent ± SEM. *p < 0.01 versus control, #p < 0.001 versus control, +p < 0.001 versus 72 hours treatment. [Click here to view] |
Multiparametric dose-response analysis
In the present study, variation in features other than potency was analyzed in two systems levels—molecular and cellular (Table 1)—in order to better understand both the response induced by these two PDK1 inhibitors and disease. Thus, at molecular system level, tests were done directly to PDK1 kinase, and the % inhibition and IC50 were evaluated, clarifying the potency of the drugs. As a result, FC100 proved to be notoriously more potent than G51. On the other hand, at the cellular system level, parameters such as GI50, TGI, HS, Einf, Emax, and AUC were evaluated to gather the biological importance of variation in other parameters than potency.
In general, GI50 values are lower for FC100 compound when compared to G51; however, the former seems not to produce a high enough perturbation to completely inhibit cell growth (TGI) up to 40 µM, indicating that IC50 and GI50 fail to indicate the most promising compound.
When analyzing AUC, their smaller values are for G51 as compared to FC100, reflecting a higher drug activity for the first one. In addition, the results obtained for TGI agree with AUC data, confirming the poor activity of this compound toward this cellular system.
 | Figure 3. Multiple comparison analysis of the G51 induced perturbation in the cellular system-level U-87 MG cell line. The bars represent the mean difference of perturbation. [Click here to view] |
Analyzing the HS parameter for G51 drug (Fig. 5; Table 1), it was found that, generally, the target in this cellular system is always fully available during the experimentation and might represent a drug with polypharmacology (HS < −1).
On the other hand, for FC100 drug (Fig. 6; Table 1), this parameter shows that a subpopulation of cells were simply unaffected and that the target of this compound is not always available in the U-87 MG cell line during the first 48 hours. However, surprisingly, at 72 hours of drug treatment, there is a steeper HS, indicating availability of drug target and some activity of the compound, also confirmed by its lowest value of AUC.
Regarding the measurement of drug efficacy for anticancer drugs, the high maximal effect is obtained when Emax ~ Einf ~ 0, which corresponds to 100% of cell death (Fallahi-Sichani et al., 2013). Analyzing our data, G51 shows closer values to 0 when compared to FC100.
In a novel drug discovery and development era, where challenges are found in every branch of the process, involving a variety of players, from the basic science, clinical, and industry, all parts must be integrated in a harmonic manner. Specifically, we are talking about of the emergent concept of Academic Drug Discovery Centres (Schultz Kirkegaard and Valentin, 2014), crowdfunding initiatives (Houghton et al., 2007), back-up strategies (Monks et al., 1991), and development of integrative analysis (Kell and Goodacre, 2014). Also, the “wet laboratory” needs to keep up with all of the updates coming from these areas and contribute actively to the improvement of this business. As a result, the purpose of this study was to develop a new way of rethinking cellular drug response and ultimately contribute for a better prediction of drug effects by exploring how the actual focus on potency ignores the potential impact of other dose-response parameters to analyze the action of two PDK1 inhibitors in GBM at the cellular system-level.
As previously reported, PKD1 is directly and indirectly involved in a variety of human cancers, specially breast cancer (Dragojlovic and Lynd, 2014; Provins et al., 2014; Schultz Kirkegaard and Valentin, 2014; Vichai and Kirtikara, 2006), but also involved in ovarian, prostate, pancreatic cancers, and glioblastoma (Dunn et al., 2012; Fyffe and Falasca, 2013; Hernandez-Aya and Gonzalez-Angulo, 2011; Kell and Goodacre, 2014; Raimondi and Falasca, 2011). In our experiments, both inhibitors perturbed the U-87MG cellular system, supporting the PDK1 aberration in GBM reported before (Dunn et al., 2012; Fyffe and Falasca, 2013). However, different effects were observed—F100 induced cell growth at 24 and 48 hours, while G51 led to cell growth arrest. An important phenomenon as polypharmacology can be the reason for the observed results since G51 and FC100 compound show different degrees of potency (Table 1), also it is supported by the HS analysis. However, the highly potent compound toward PDK1 inhibition (FC100) does not induce a specific anticancer effect on the U-87MG cellular system, although having some nonspecific activity after 72 hours of treatment, while G51 shows a cytostatic effect. This indicate two important things: first, the cytostatic effect shown by G51 might not be related exclusively to PDK1 inhibition; second, the parameter potency cannot be used to exclusively to stratify drugs during the R&D process.
Regarding the first conclusion, it has been shown by literature that PDK1 controls migration and malignant transformation but not cell growth and proliferation in PTEN-null lymphocytes (Dragojlovic and Lynd, 2014). It is also reported that in breast cancer, PDK1 just plays an essential role in regulating cell migration (Provins et al., 2014), reinforcing our results regarding the lack of growth inhibition effect or cell killing on this cell line. Therefore, in a therapeutic context, PDK1 inhibitors possibly play a role in combination with other antitumor agents to treat glioblastoma multiforme. This conclusion is supported by the literature already published: PDK1 and checkpoint kinase 1 (CHK1) inhibition is required to induce antitumor effect in GBM in vivo by killing glioblastoma stem-like cells (Sargeant et al., 2007), as well as, another study states that by reversing the Warburg Effect, combined targeting of PDK1 and epidermal growth factor receptor inhibited glioblastoma multiforme growth and proliferation (Fyffe and Falasca, 2013).
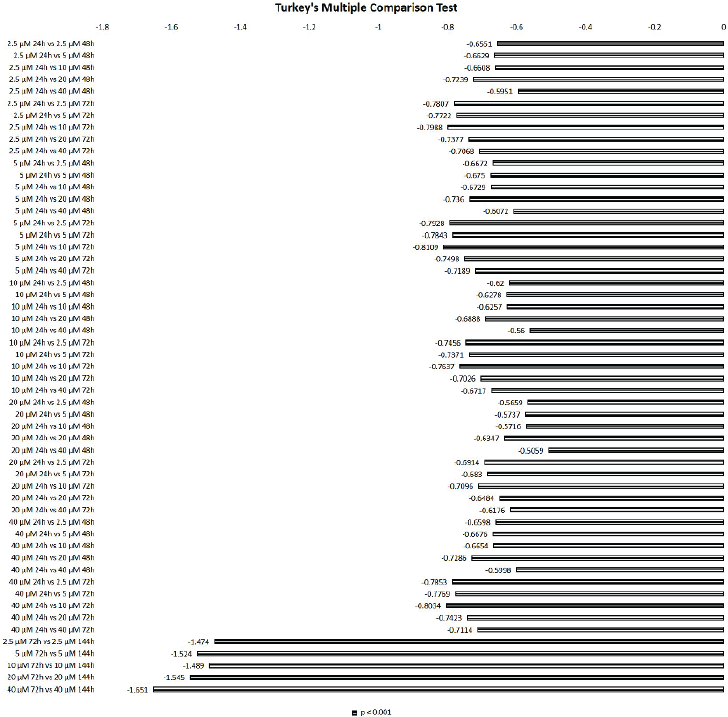 | Figure 4. Multiple comparison analysis of the FC100 induced perturbation in the cellular system-level U-87 MG cell line. The bars represent the mean difference of perturbation. [Click here to view] |
Talking about the parameter potency, it fails to indicate our lead compound to treat glioblastoma multiforme. Therefore, this suggests that it is not the most important parameter to characterize a cellular response, and so, to stratify drugs at early stage of the drug discovery process, as it has been done along these years. Inclusively, other parameters that did not receive too much attention seem to emerge now in our study as better predictors to stratify compounds. For example, HS starts to gain more attention on the literature (Guven-Maiorov et al., 2014; Jenkins, 2013). Also, regarding our results, the shape of dose-response curves is affected by polypharmacology and further analysis of G51 compound throughout a kinase profile and at the proteomics level will confirm it.
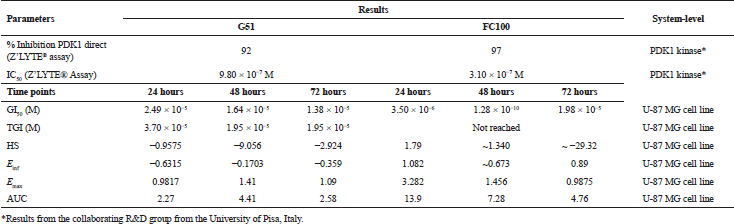 | Table 1. Multiparametric concentration-response parameters for G51 and FC100 compounds. [Click here to view] |
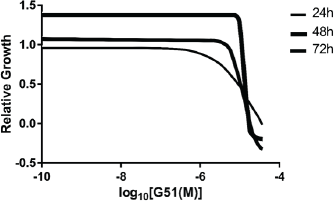 | Figure 5. Dose-response curves for G51 showing normal (HS = −1), steep (HS < −1) or shallow (HS > −1) responses for each curve. [Click here to view] |
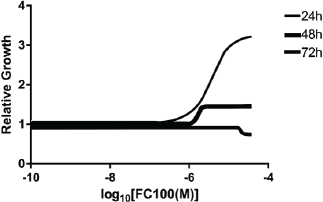 | Figure 6. Dose-response curves for FC100 showing normal (HS = −1), steep (HS < −1) or shallow (HS > −1) responses for each curve. [Click here to view] |
CONCLUSION
Overall, this study suggests that in this cell line, analyzing other parameters seems to not yield extra information when it comes to characterize drugs profile at the cellular system level as in the end, those parameters correlated with each other. However, here both compounds tested in U-87MG, HS gathered the same information as the % inhibition PDK1 direct performed by Z’LYTE® Assay at the University of Pisa, because, as we see, the compound that most inhibits PDK1 is the one that does not show effect. Therefore, the results from both assays agree when it comes to the polypharmacology of such compound. So, HS should be further explored as a marker that yields polypharmacology since a single procedure at the cellular level could clarify both drug profiles and cellular response, saving the molecular studies to reveal and confirm the multiple drug targets and so, saving time & reducing costs.
Finally, taking in account that over 50% of failures at phases II and III clinical trials are attributed to a lack of efficacy, which could be a result of a no optimal drug target (Velpula and Tsung, 2014), another question must be done: is PDK1 an optimal target to glioblastoma multiforme? In our study, PDK1 inhibition does not always result in GBM growth inhibition in this cell line. Therefore, further system-level studies in other cell lines and in other systems, including at protein level, organism level, and other molecular genetics studies should be done to clarify the relevance of this target and its validity to treat glioblastoma multiforme.